Autor: yolandamupu
ANSIEDAD
La ansiedad es una emoción que surge cuando una persona se siente en peligro, sea real o imaginaria la amenaza. Es una respuesta normal o adaptativa, que prepara al cuerpo para reaccionar ante una situación de emergencia. Por lo tanto, tiene una función muy importante relacionada con la suervivencia, junto con el miedo, la ira, la tristeza o la felicidad. Para preservar su integridad física ante amenazas, el ser humano ha tenido que poner en marcha respuestas eficaces y adaptativas durante millones de años: la reacción de lucha-huida.

Ante una situación de alerta, el organismo pone a funcionar el sistema adrenérgico. Por ejemplo, cuando el organismo considera necesario alimentarse, este sistema entra en funcionamiento y libera señales de alerta a todo el sistema nervioso central. Cuando se detecta una fuente de alimento para la cual se requiere actividad física, se disparan los mecanismos que liberan adrenalina, y se fuerza a todo el organismo a aportar energías de reserva para la consecución de una fuente energética muy superior a la que se está invirtiendo para conseguirla y que normalizará los valores que han disparado esa «alerta amarilla». En esos momentos el organismo, gracias a la adrenalina, pasa a un estado de «alerta roja».

El sistema dopaminérgico también se activa cuando el organismo considera que va a perder un bien preciado. En esta situación, el organismo entra en alerta amarilla ante la posibilidad de la existencia de una amenaza, que no es lo mismo que cuando la amenaza pasa a ser real, pues en ese caso lo que se libera es adrenalina.

Desde este punto de vista, la ansiedad se considera una señal positiva, de salud, que ayuda en la vida cotidiana, siempre que sea una reacción frente a determinadas situaciones que tengan su cadena de sucesos de forma correlativa: alerta amarilla, alerta roja y consecución del objetivo. Si la cadena se rompe en algún momento y esas situaciones se presentan con ansiedad, entonces el organismo corre el riesgo de intoxicarse por dopaminas o por otras catecolaminas. Esas situaciones ayudan al organismo a resolver peligros o problemas puntuales de la vida cotidiana.

En las sociedades avanzadas modernas, esta característica innata del ser humano se ha desarrollado de forma patológica y conforma, en algunos casos, cuadros sintomáticos que constituyen los denominados trastornos de ansiedad, que tiene consecuencias negativas y muy desagradables para quienes lo padecen. Entre los trastornos de ansiedad se encuentran las fobias, el trastorno obsesivo-compulsivo, el trastorno de pánico, la agorafobia, el trastorno por estrés postraumático, el trastorno de ansiedad generalizada, el trastorno de ansiedad social, etc. El miedo escénico es una forma de ansiedad social, que se manifiesta frente a grupos y ante la inminencia de tener que expresarse en público o por efecto de imaginar dicha acción. En el caso del trastorno de ansiedad generalizada, la ansiedad patológica se vive como una sensación difusa de angustia o miedo y deseo de huir, sin que quien lo sufre pueda identificar claramente el peligro o la causa de este sentimiento. Esta ansiedad patológica es resultado de los problemas de diversos tipos a los que se enfrenta la persona en su vida cotidiana, y sobre todo de sus ideas interiorizadas acerca de sus problemas.

No se conocen totalmente las causas de los trastornos de ansiedad, pero se sabe que la interacción de múltiples determinantes favorece su aparición. Se conoce la implicación tanto de factores biológicos como ambientales y psico-sociales. Además, es muy común la comorbilidad con otros trastornos mentales, como los trastornos del estado de ánimo.

Entre los factores biológicos, se han encontrado alteraciones en los sistemas neurobiológicos gabaérgicos y serotoninérgicos; anomalías estructurales en el sistema límbico (córtex paralímbico), que es una de las regiones más afectadas del cerebro; ciertas alteraciones físicas; una mayor frecuencia de uso y/o retirada de medicinas, alcohol, drogas y/o sedantes y otras sustancias; y cierta predisposición genética.

Entre los factores ambientales, se ha encontrado la influencia de ciertos estresores ambientales, una mayor hipersensibilidad y una respuesta aprendida. Los factores psicosociales de riesgo son las situaciones de estrés, las experiencias que amenazan la vida, el ambiente familiar y las preocupaciones excesivas por asuntos cotidianos. Determinadas características de la personalidad pueden ser factores predisponentes.

La ansiedad normal es adaptativa y permite a la persona responder al estímulo de forma adecuada. Se presenta ante estímulos reales o potenciales (no imaginarios o inexistentes). La reacción es proporcional cualitativa y cuantitativamente, en tiempo, duración e intensidad.

La ansiedad se considera patológica cuando el estímulo supera la capacidad de adaptación de respuesta del organismo y aparece una respuesta no adaptativa, intensa y desproporcionada, que interfiere con el funcionamiento cotidiano y disminuye el rendimiento. Se acompaña de una sensación desagradable y desmotivadora, síntomas físicos y psicológicos, y persiste más allá de los motivos que la han desencadenado.
La ansiedad patológica presenta las siguientes características: se manifiesta intensamente, se prolonga y mantiene en el tiempo más de lo debido, aparece de forma espóntanea sin un estímulo desencadenante (de manera endógena), surge ante estímulos que no debieran generar la respuesta de ansiedad y se presenta una respuesta inadecuada respecto al estímulo que lo suscita.

El límite entre la ansiedad normal y la ansiedad patológica no es fácil de definir y puede variar entre los individuos en función de los rasgos de personalidad o, sobre todo, en función de lo que se ha descrito como un «estilo cognitivo propenso a la ansiedad». Los criterios diagnósticos del Manual diagnóstico y estadístico de los trastornos mentales, ediciones cuarta y quinta (DSM-IV y DSM-5, respectivamente), señalan que la ansiedad debe considerarse patológica cuando «La ansiedad, la preocupación o los síntomas físicos provocan malestar clínicamente significativo o deterioro social, laboral o de otras áreas importantes de la actividad.» Es útil distinguir entre la ansiedad «estado», que es episódica y transitoria, y la ansiedad «rasgo», que es persistente y puede reflejar una personalidad «propensa a la ansiedad».
Si una persona reacciona en alguna ocasión con altos niveles de ansiedad ante una situación, ante la que otras no experimentan tanta ansiedad, se puede considerar simplemente una reacción de alta intensidad, o aguda en un nivel no demasiado alto, que es puntual y no extrema. Esto no suele suponer ningún trastorno.
El problema surge cuando esta forma de reacción aguda es excesivamente intensa, como en los ataques de pánico o en las crisis de ansiedad (en los que la persona no puede controlar su ansiedad y alcanza niveles extremos), o bien cuando dicha reacción aguda se establece como un hábito, es decir, si una reacción de ansiedad de alta intensidad se convierte en crónica, o se vuelve muy frecuente.
Una reacción aguda de ansiedad no siempre es patológica, sino que puede ser muy adaptativa. Por ejemplo, cuando la situación que la provoca requiere una fuerte reacción de alarma que prepare para la acción (si se exige una gran concentración en una tarea para la que se necesitan muchos recursos de la atención); o si requiere una gran activación a nivel fisiológico (porque se necesita tensar más los músculos, bombear mayor cantidad de sangre, más oxígeno, etc.). Dicha reacción de ansiedad ayuda a responder mejor ante esta situación
El primer paso ante un paciente con síntomas de ansiedad es realizar una completa evaluación, que puede incluir diversas pruebas adicionales, para excluir o confirmar la presencia de una causa orgánica subyacente o asociada, que esté provocando los síntomas de ansiedad. Para ello, se tienen en cuenta los síntomas físicos que predominan, la historia médica y psicológica previa tanto del paciente como de su familia y las enfermedades que generan trastornos de ansiedad, así como la probabilidad de que las pueda padecer.

Existe un amplio abanico de enfermedades que cursan con síntomas psiquiátricos o que pueden simular un trastorno mental. Su identificación puede llegar a resultar complicada y no siempre se realiza una adecuada evaluación del paciente.

En ocasiones, los síntomas psiquiátricos se desarrollan antes de la aparición de otros síntomas o signos más característicos de la enfermedad, como ocurre en ciertos trastornos metabólicos, e incluso pueden ser las únicas manifestaciones de la enfermedad en ausencia de cualquier otro síntoma, como ocurre en algunos casos de enfermedad celíaca o de sensibilidad al gluten no celíaca, por lo que con frecuencia no se consigue un diagnóstico correcto o este se demora durante años.
Algunos de los trastornos que cursan frecuentemente con síntomas de ansiedad incluyen:
• Trastornos endocrinos, tales como el hipotiroidismo, el hipertiroidismo, la hiperprolactinemia, la psicosis posparto o el síndrome de Cushing.
• Enfermedades sistémicas, inflamatorias o infecciosas, tales como la enfermedad celíaca y la sensibilidad al gluten no celíaca (ambas cursan con frecuencia sin síntomas digestivos), el lupus eritematoso sistémico, el síndrome antifosfolípidos, la mononucleosis infecciosa, la sepsis, la fiebre tifoidea, la brucelosis, la malaria, la enfermedad de Lyme o el VIH/sida.
• Alergias.
• Enfermedades gastrointestinales, tales como la enfermedad inflamatoria intestinal.
• Estados carenciales, por déficit de vitaminas B2, B12, D o ácido fólico.
• Trastornos electrolíticos o de fluidos, tales como la hiponatremia o la hipocalcemia.
• Fallo hepático, como la encefalopatía hepática.
• Fallo renal, como la retención urinaria aguda.
• Enfermedades respiratorias, tales como el asma, el edema pulmonar, la embolia pulmonar, el trasplante de pulmón, la enfermedad pulmonar obstructiva crónica (EPOC), el mal de altura o la hipoxemia.
• Trastornos metabólicos, tales como la hipoglucemia o la hiperglucemia.
• Enfermedades cardíacas, tales como las arritmias cardíacas, la insuficiencia cardíaca, la enfermedad de las arterias coronarias, el prolapso de la válvula mitral o el trasplante de corazón.
• Enfermedades hematológicas, tales como la anemia, la policitemia, la leucemia o la anemia de células falciformes.
• Trastornos neurológicos, tales como la enfermedad de Alzheimer, la demencia vascular, la enfermedad de Huntington, la enfermedad de Parkinson, la esclerosis múltiple, la enfermedad de Wilson, los tumores cerebrales, los accidentes vasculares cerebrales, las enfermedades vasculares cerebrales crónicas o la hidrocefalia.
• Enfermedades infecciosas del cerebro, tales como la meningitis, la encefalitis o la neurosífilis.
• Consumo de sustancias tóxicas, como la cafeína, el cannabis o la cocaína y otras drogas de síntesis. Asimismo, muchas de las personas que padecen ansiedad (sobre todo ansiedad generalizada, trastorno de angustia y fobia social), consumen alcohol con el pretendido objetivo de aliviar la sintomatología de la angustia.
• La evolución de los problemas de ansiedad cursa con períodos de reducción y desaparición de los síntomas durante un intervalo de tiempo variable. De la misma forma que ocurre con cualquier otra enfermedad crónica, con un tratamiento apropiado se puede convivir con este problema de manera adecuada, consiguiendo llevar una vida normal. Un tratamiento efectivo ayuda a disminuir los síntomas, mejorar la autoestima, volver a disfrutar de la vida de nuevo y prevenir recaídas, si bien pueden aparecer altibajos durante el proceso.
• Los tratamientos habituales son la psicoterapia (terapia cognitivo-conductual) y la medicación (principalmente antidepresivos y ansiolíticos), que pueden ser usados o no de forma conjunta, según el trastorno que presente el paciente.

Según la psicología cognitiva, los pensamientos generados por la ansiedad «producen distorsiones a la hora de orientarse en el mundo» y mirar la realidad.
• 1. Pesimismo: «tendencia a focalizarse en el problema sin ser capaz de ver las soluciones».
• 2. Generalización: «los pensamientos son tipo siempre/nunca, todo/nada».
• 3. Pensamiento negativo: «el foco está en los aspectos negativos y se olvidan o descalifican los positivos».
• 4: Catastrofismo: «ver los aspectos negativos de una manera excesiva y exagerada».
• 5. Leer el pensamiento: «creen saber lo que los otros están pensando y sus motivos negativos ocultos».
• 6. Adivinar el futuro: «tendencia a anticipar que las cosas van a salir mal».
• 7. Comparación: «medirse con los demás para acabar siempre perdiendo y sintiéndose inferior».
• 8. Exageración: «si alguien se equivoca una vez pasa a ser un torpe o si le sale mal una cosa le llama fracasado en todas las áreas».
• 9. Culpabilidad: «sentir que las circunstancias desagradables que suceden siempre están en relación con uno mismo»
• 10. Perfeccionismo: «establecer exigencias a los demás, a uno mismo o a cómo deberían ser las cosas».

AUTISMO Y TRASTORNOS DEL ESPECTO AUTISTA.
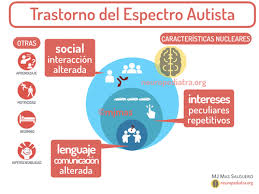
Los trastornos del espectro del autismo o TEA abarcan un amplio espectro de trastornos que, en su manifestación fenitípica, se caracterizan por deficiencias persistentes en la comunicación social y en la interacción social en diversos contextos, unidas a patrones restrictivos y repetitivos de comportamiento, intereses o actividades. Estos síntomas han de estar presentes en las primeras fases del período de desarrollo de la persona, aunque pueden no manifestarse totalmente hasta que las demandas sociales superan sus limitaciones. También pueden permanecer enmascarados por estrategias aprendidas. La historia del estudio científico del autismo comienza con la publicación en 1943 del artículo «Autistic disturbances of affective contact» («Trastornos autistas del contacto afectivo») de Leo Kanner (1943), pero sufrirá diversos avatares que retrasarán el avance de la investigación hasta bien entrado el decenio de 1960.

Durante mucho tiempo, el autismo fue considerado un trastorno infantil. Sin embargo, hoy día se sabe que se trata de una condición permanente que acompaña a la persona a lo largo de todo su ciclo vital. Aunque aún no está clarificada su etiología, los trastornos del espectro autista parecen estar causados por la interacción entre una susceptibilidad genética heredable y factores epigenéticos y ambientales que actúan durante la embrogenesis.

Las características de los trastornos de especto autista son:
1. Déficit en la reciprocidad socio-emocional, que oscilan desde un acercamiento social inadecuado y errores en el seguimiento de una conversación; un nivel reducido de compartir intereses, emociones o afectos; fracaso para iniciar o responder a las interacciones sociales.
2. La falta de atención a partir del año y medio,aparentemente no escucha.
3. Déficit en las conductas de comunicación no verbal empleados para la interacción.
4. Déficit en el desarrollo, mantenimiento y comprensión de las relaciones; que abarcan; por ejemplo, desde las dificultades para ajustar su conducta para adaptarse a varios contextos sociales; dificultades para compartir el juego imaginativo o para hacer amigos, hasta la ausencia de interés por los iguales.

La gravedad está basada en las deficiencias en la comunicación social y en los patrones de conducta restrictivos o repetitivos.
El diagnóstico del TEA es con frecuencia un proceso de dos etapas. La primera etapa comprende una evaluación del desarrollo general durante los controles del niño sano con un pediatra. Los niños que muestran algunos problemas de desarrollo se derivan para una evaluación adicional. La segunda etapa comprende una evaluación exhaustiva efectuada por un equipo de médicos y otros profesionales de la salud con un amplio rango de especialidades. En esta etapa, un niño puede recibir un diagnóstico de autismo o de algún otro trastorno del desarrollo.

Mucha gente—inclusive pediatras, médicos de familia, maestros y padres—pueden, al principio, ignorar los signos del TEA, al creer que los niños «alcanzarán» a sus compañeros. Aunque a usted pueda preocuparle pensar que su hijo pequeño tiene el TEA, cuanto más temprano se diagnostique el trastorno, más rápidamente pueden comenzar las intervenciones.
Un control del niño sano debería incluir una prueba para evaluar su desarrollo, con examen de detección específico del TEA a los 18 y 24 meses, como lo recomienda la Academia Americana de Pediatría. Realizar exámenes de detección del TEA no es lo mismo que diagnosticar el TEA.
A veces, el médico interrogará a los padres acerca de los síntomas del niño a fin de detectar el TEA. Otros instrumentos de detección combinan información de los padres con observaciones del niño realizadas por el médico. Los ejemplos de instrumentos de detección para los infantes y niños en edad preescolar incluyen:
• Lista de verificación para el autismo en los infantes (CHAT, por sus siglas en inglés)
• Lista de verificación modificada para el autismo en los infantes (M-CHAT, por sus siglas en inglés)
• Herramienta de detección del autismo en niños de dos años (STAT, por sus siglas en inglés)
• Cuestionario de comunicación social (SCQ, por sus siglas en inglés)
• Escalas de conducta comunicativa y simbólica (CSBS, por sus siglas en inglés).
• Para detectar el TEA leve o síndrome de Asperger en los niños mayores, se puede depender de instrumentos de detección diferentes, como:
• Cuestionario de exploración del espectro autista (ASSQ, por sus siglas en inglés)
• Escala australiana para el síndrome de Asperger (ASAS, por sus siglas en inglés)
• Test infantil del síndrome de Asperger (CAST, por sus siglas en inglés).

La segunda etapa de diagnóstico debe ser minuciosa a fin de encontrar si otras afecciones pueden ser las causantes de los síntomas del niño. Un equipo que incluye un psicólogo, un neurólogo, un psiquiatra, un logopeda u otros profesionales experimentados en el diagnóstico del TEA puede efectuar esta evaluación. La evaluación puede calificar el nivel cognitivo del niño, el nivel de lenguaje, su conducta adaptativa (habilidades adecuadas en relación con la edad necesaria para completar las actividades diarias independientemente, por ejemplo, alimentarse, vestirse y asearse), los niveles de audición, imágenes cerebrales y exámenes genéticos.
Los datos, pues, apuntan a que el fenotipo autista es independiente de la inteligencia. Es decir, se pueden encontrar autistas con cualquier nivel de inteligencia. Aquellos con inteligencia por debajo de lo normal serían los que tienden a ser diagnosticados. Aquellos con inteligencia normal o superior suelen escapar al diagnóstico.
El autismo infantil produce alteraciones intelectuales que a menudo son muy difíciles de diferenciar del discapacidad intelectual. Sus principales características son:
• Ausencia de interacción social
• Alteraciones profundas en el lenguaje, no acorde con las capacidades intelectuales
• Insistencia en comportamientos estereotipados
• Aparece antes de los 30 meses de edad
• Resistencia al cambio
• Incapacidad para anticipar el peligro
Su cociente intelectual suele ser bajo, correlacionándose en forma directa con los defectos lingüísticos. En pruebas psicométricas, el perfil de inteligencia del niño autista (al contrario del menor con retraso mental) con frecuencia muestra:
• Disociación entre los CI verbal y no verbal, con una superioridad por parte de las habilidades no verbales
• El desarrollo del lenguaje no sigue las etapas normales
• Regresiones espontáneas en el proceso de desarrollo comunicativo
• Disociaciones claras entre la forma y el contenido del lenguaje y su uso en forma inapropiada

Los menores que padecen retraso mental suelen exhibir un retraso en el desarrollo lingüístico, pero siguen las mismas etapas del niño normal. El autismo infantil y el retraso mental llegan a estar relacionados y, de hecho, se ha considerado que aproximadamente tres cuartas partes de niños autistas funcionan como adultos con retraso mental.

Los objetivos de las intervenciones son disminuir el impacto de los déficits tanto en la vida personal como en la familiar y social, mejorar la calidad de vida y la independencia funcional. Ningún tratamiento se ha establecido como superior y generalmente debe ser adaptado a las necesidades del niño.

Los programas de educación especial, intensiva y sostenida, y las cognitivo conductuales en etapas tempranas de la vida han mostrado su eficacia para ayudar a adquirir habilidades de cuidado personal, sociales y de trabajo. Los programas de intervención temprana (de 0 a 6 años de edad) han demostrado su eficacia en la contención o eliminación de síntomas autísticos, en mejoras perceptivas, de atención, cognitivas, comunicativas o de las habilidades sociales. Es necesario, además, que la intervención se lleve a cabo con una perspectiva holística, e incidir de manera interdisciplinaria sobre todos los aspectos que ofrezcan disfunciones, bien sea en la conducta social, en el manejo de la comunicación y del lenguaje o en el comportamiento. Se trata de mejorar la situación del niño con TEA y sus habilidades, pero al mismo tiempo su bienestar, su calidad de vida y la de su familia.

Por un lado, la Tecnología Educativa (TE) es el conjunto de conocimientos, aplicaciones y dispositivos o herramientas que otorgan la posibilidad de aplicar las TIC en el ámbito educativo, concretamente en los procesos de enseñanza y aprendizaje. Gracias a ésta, los docentes pueden diseñar y planificar el proceso de enseñanza y aprendizaje de manera óptima y eficiente.

Por otro lado, las Necesidades Específicas de Apoyo Educativo (NEAE) engloban todas las Necesidades Educativas Especiales (NEE) y Necesidades Educativas que pueda tener el alumnado. Estas últimas se derivan de Dificultades Específicas de Aprendizaje (DEA), del Trastorno de déficit de Atención con o sin hiperactividad (TDAH), de las Altas Capacidades Intelectuales, de Especiales Condiciones Personales o de Historia Escolar (ECOPHE), o bien por Incorporación Tardía al Sistema Educativo.

La Tecnología Educativa dirigida a las personas afectadas con autismo se le conoce como «tecnología asistente» que sirve principalmente para aprender habilidades sociales y a comunicarse con los demás, marcar rutinas o bien identificar situaciones cotidianas. Para considerarlas útiles estas herramientas han de cumplir los siguientes requisitos:
• fomentar el aprendizaje y ser intuitivas, es decir, fáciles de usar.
• deben ser flexibles, adaptables y personalizables.
• Ser amenas, atractivas y motivadoras.

El pronóstico del autismo es aparentemente impredecible. Algunos niños se desarrollan a niveles en los cuales su autismo no es comúnmente perceptible, sin razón aparente. Otros desarrollan habilidades funcionales después de un tratamiento intenso con terapia ABA. Algunos padres reportan mejorías después de utilizar tratamientos biológicos (no probados). Por otro lado, muchos individuos autistas requieren ser cuidados de por vida y otros nunca desarrollan lenguaje oral. La terapia parece no tener efecto alguno en ciertos casos. Mientras que algunos autistas adultos parecen mejorar en su funcionamiento al pasar el tiempo, otros reportan que se vuelven «más autistas».

La ansiedad y la depresión se presentan con frecuencia en adolescentes y adultos autistas. Se sabe que la respuesta al estrés es más pronunciada en muchos autistas, lo cual podría ser una causa. Pero dados los déficits sociales de los autistas, también es posible que la ansiedad y depresión se deban a instancias de adversidad social.
Los niños autistas pueden asistir a escuelas regulares, siempre y cuando cuenten con los apoyos que requieren para aprender y desarrollarse en la escuela. Cada niño es único con sus fortalezas, gustos y retos. Es decir que tampoco los niños con autismo son iguales entre sí, por lo que en la escuela se debe formar un equipo de trabajo junto con la familia y si es necesario especialistas externos. Este equipo se encarga de definir los objetivos para el alumno, así como la forma en que van a trabajar con él. Es muy importante tomar en cuenta las fortalezas del niño al diseñar su programa.

Es así mismo de vital importancia crear conciencia en los colegios y escuelas acerca del autismo y sus variantes (como el síndrome de Asperger, por ejemplo) a fin de erradicar el acoso escolar o bullying del cual pueden ser víctimas a causa de la ignorancia.

Para las personas con discapacidad las dimensiones de la calidad de vida son semejantes a las del resto de la población.
• La calidad de vida mejora en el momento en el que las personas con esta condición se sienten con el poder de tomar decisiones que repercutan en su vida, durante muchos años este poder se les ha quitado de las manos a las personas con discapacidad, siendo asumido por familiares o los profesionales cercanos a la persona.
• La calidad de vida aumenta con la aceptación y la total integración de las personas a la comunidad en la que se encuentran, brindándoles independencia, siendo esta un factor esencial en la percepción de la calidad de vida.
• Todas las personas experimentamos la calidad de vida en el momento en el que las necesidades básicas se cumplen y se dan las mismas oportunidades para lograr distintas metas en contextos tales como hogar, comunidad, escuela y trabajo.

Un criterio común para la distinción entre autismo de alto y de bajo funcionamiento es un cociente intelectual de más de 70-80 para aquellos que se dice que son de alto funcionamiento, y de menos de 70-80 para aquellos que se dice que son de bajo funcionamiento. Este criterio tiene varios problemas:
• Se cree que las pruebas de cociente intelectual son inadecuadas para medir la inteligencia de una persona autista, ya que están diseñadas para personas típicas. Es decir, estas pruebas asumen que existe interés, entendimiento, conocimientos lingüísticos, motivación, habilidad motriz, etc. Se conocen casos de personas autistas cuyo cociente intelectual cambia drásticamente dentro de un periodo relativamente corto, lo cual probablemente no indica un cambio real en el nivel de inteligencia.
• La percepción de «bajo funcionamiento» por lo general se refiere a carencia de habla, incapacidad para cuidarse de sí mismo, falta de interacción social, etc. Esto no siempre coincide con el criterio del cociente intelectual. Existen personas autistas que carecen de habla (aunque se pueden comunicar por escrito) con un cociente intelectual alto. Por otro lado, autistas con un cociente intelectual bajo podrían poseer la capacidad del habla.
• Los autistas varían extremadamente en sus capacidades. Una misma persona puede mostrar características de «alto funcionamiento» y otras de «bajo funcionamiento.» Por lo tanto estas etiquetas son uni-dimensionales y su descriptividad deficiente.
• Las personas autistas que son de «bajo funcionamiento» en algún área pueden desarrollarse y volverse de «alto funcionamiento» en esa misma área. Alguien diagnosticado autista puede volverse indistinguible de alguien diagnosticado con Síndrome de Asperger.
El Día del Orgullo Autista se celebra cada 18 de junio desde 2005. En España, el 18 de noviembre de 2014, con el respaldo de todos los grupos parlamentarios, se aprobó una proposición no de ley en la que se instaba al Gobierno a estudiar, en el ámbito de sus competencias, la elaboración y desarrollo de una Estrategia Española en Trastornos del Espectro del Autismo.

DIARIO DE SORIA: VIH

VIH
El virus de la inmunodeficiencia humana (VIH) es un lentivirus(un género de la familia retrovirus) que causa la infección por VIHy con el tiempo el síndrome de inmunodeficiencia adquirida(sida). El sida es una enfermedad humana que progresa hacia la falla del sistema inmune lo que permite que se desarrollen infecciones importunistas y cánceres potencialmente mortales. Sin tratamiento, se estima que la sobrevida promedio después de la infección de VIH es de nueve a once años; dependiendo en el subtipo de VIH. La infección por VIH ocurre únicamente a través de los siguientes fluidos de personas infectadas: sangre, semén, flujo vaginal, líquido preseminal y leche de lactancia. Dentro de estos fluidos corporales, el VIH está presente tanto como partículas libres y virus dentro de células inmunes infectadas.

El VIH infecta células vitales en el sistema inmune humano como las células T helper (específicamente células CD4+), macrofagos y células dentriticas. La infección por VIH puede llevar a niveles bajos de células T CD4+ a través de varios mecanismos, incluidos la piroptosisde células T infectadas inutilizadas, apoptosis de células no infectadas próximas, muerte viral directa de las células infectadas y muerte de las células T CD4+ por los linfocitos citotóxicos CD8 que reconocen a las células infectadas. Cuando el número de células T CD4+ disminuyen bajo un nivel crítico, se pierde la inmunidad celular y el organismo se vuelve progresivamente más susceptible a las infecciones oportunistas.
Categorización
El virus de la inmunodeficiencia humana forma parte del genero lentivirus. Estos constituyen un grupo dentro de la familia Retroviridae. Los virus de este grupo poseen propiedades morfológicas y biológicas comunes. Varias especies son atacadas por los lentivirus, cuya característica principal consiste en un período de incubación prolongado que desemboca en enfermedad después de varios años.
El VIH fue descubierto y considerado como el agente de la naciente epidemia de sida por el equipo de Luc Montanier en Francia en 1983. El virion es esférico, dotado de una envoltura y con una cápside proteica. Su genoma es una cadena de ARN monocatenario que debe copiarse provisionalmente al ADN para poder multiplicarse e integrarse en el genoma de la célula que infecta. Los antígenos proteicos de la envoltura exterior se acoplan de forma específica con proteínas de la membrana de las células infectables, especialmente de los linfocitos T CD4.

Desde su ingreso en la célula hospedadora, la cadena simple de ácido ribonucleico (ARN) viral comienza su transformación en una doble cadena de ácido desoxirribonucleico (ADN) por acción de la encima trascriptasa inversa que forma parte del virus. La integrasa y otros cofactores actúan para que el ARN del virus se fusione con el ADN de la célula hospedadora a través de la trascripcion en el genoma de la célula que aloja al virus. De esta manera, la célula queda infectada por el virus. Después de este proceso, los lentivirus reaccionan de dos maneras posibles: puede ocurrir que el virus entre en latencia mientras la célula infectada continúa en funciones, o bien, que el virus comience a replicarse activamente y libere viriones capaces de infectar otras células.

Existen dos tipos del VIH, llamados VIH-1 y VIH-2. El primero de ellos corresponde al virus descubierto originalmente, que recibió los nombres de LAV y HTLV-III por parte de los dos equipos que estaban investigando el agente etiológico del sida durante la primera mitad de la década de 1980. El VIH-1 es más virulento e infeccioso que el VIH-2 y es el causante de la mayoría de infecciones por VIH en el mundo. El VIH-2 es menos infeccioso y se encuentra confinado casi exclusivamente a los países de Africa Occidental.
Estructura y genoma del VIH
Estructura
El VIH comparte con los retrovirus las características esenciales de esa familia. El virión contiene información genética bajo la forma de ácido ribonucleico (ARN), protegido por una envoltura de membrana. Los retrovirus insertan su información genética en las células hospedadoras por acción de la trascriptasa inversa.
Un virión del VIH tiene una forma aproximadamente esférica con un diámetro de 80-100 nm. Está constituido por tres capas. La exterior es una bicapa lipídica. Posee 72 espículas formadas por las glicoproteínas gp120y gp41 que actúan en el momento de la unión del virus a la célula hospedadora. La capa intermedia está constituida por la núcleo cápside icosaedrica. La capa interior tiene forma de un cono truncado. Está constituida por el ARN viral y la nucleoproteína. La cadena genética del VIH está constituida por un ARN de cadena simple compuesto por dos filamentos idénticos. El ARN contiene varios genes, cada uno de los cuales codifica las diversas proteínas que el VIH necesita para reproducirse.

Genoma y composición
Los genomas del VIH-1 y VIH-2 son muy similares. Ambos están compuestos por los tres genes básicos de la familia de los retrovirus. Se trata de los genes gag, pol y env. Cada uno de estos genes codifica proteínas que ayudan a la reproducción del virus. El genoma del VIH posee otros seis genes adicionales: tat, vpr, rev, vpu (vpx en el caso del VIH-2), vif y nef.
Genes estructurales
Las proteínas estructurales son codificadas por los genes gag, pol y env, y su secuencia cubre la mayor parte del genoma viral, quedando sólo una parte menor para el resto de los genes.

El gen gag es traducido a una proteína precursora, la p55, que luego se asocia, durante la gemación por la que se liberan nuevas partículas víricas desde la célula infectada, a dos copias del ARN viral, para el que presenta una región afín, y a otras proteínas virales y celulares. Una proteasa, producto del gen pol corta durante la maduración del virión la p55 en cuatro proteínas que se incorporan a sus lugares respectivos:
• La proteína p24 forma la cápside.
• La proteína p17 constituye la matriz, situada bajo la envoltura, a la que estabiliza. Una parte de las proteínas se unen al complejo molecular que acompaña al ADN viral al interior del núcleo. En la superficie de la proteína existe una región cariofílica (literalmente afín al núcleo) que es reconocida por la maquinaria molecular de importación nuclear. Éste es el mecanismo que permite al VIH infectar células diferenciadas, no destinadas a dividirse, algo que no ocurre en ningún otro retrovirus.
• Las proteínas p6 y p7 (ó p9) forman la nucleocápside. La región de la p55 correspondiente al polipéptido p6 es responsable de la incorporación de la proteína accesoria Vpr (producto de la traducción del gen vpr) al virión en formación y de la interacción con la membrana de la célula que hace posible la gemación. La p7 (p9) es responsable del reconocimiento y la incorporación del ARN al virión y además interviene en la transcripción inversa facilitándola.
Dentro de la cápside, además de las dos copias idénticas del ARN viral hay ejemplares de tres enzimas necesarias para la multiplicación del virus: una transcriptasa inversa, una integrasa y una proteasa. Estas enzimas, así como una ARNasa se producen a partir de la proteína Pol, después del corte de una proteína precursora mixta derivada de la cotraducción, una de cada 20 veces, de los genes gag y pol. La propia proteasa vírica rompe la proteína anterior, con una eficiencia limitada, para obtener las proteínas Gag (p55) y Pol. Luego la proteína precursora Pol es cortada a su vez para formar las cuatro proteínas funcionales citadas:
• La proteasa (p10). Se trata de una aspartil-proteasa cuya forma funcional es un dímero del que se conoce la estructura tridimensional. Actúa cortando las piezas de las proteínas Gag, Pol y de la Gag-Pol. Una parte de los fármacos empleados contra el VIH son inhibidores de su función

• La trascrptasa inversa (p50) cuya función es la síntesis del ADN de doble cadena del provirus usando como patrón la cadena singular del ARN viral. Es una ADN-polimerasa que puede actuar como dependiente del ADN tanto como del ARN. Una vez formada la primera cadena de ADN, complementaria del ARN viral, la ARNasa lo separa de él, lo que permite a la transcriptasa inversa ejecutar la síntesis de la segunda cadena de ADN tomando como molde la primera que se formó. Así pues, para la síntesis de la primera cadena la actividad de la transcriptasa inversa es ARN-dependiente, pero para la de la segunda es ADN-dependiente. También existen múltiples fármacos contra la actividad de la trascriptasa inversa

• La ARNasa (p15), que como se ha dicho separa las cadenas de ARN de las de la ADN durante la transcripción inversa.
• La integrasa (p31) realiza la inserción del ADN proviral en el genoma de la célula huésped. No se requiere ATP para su actividad y debe cumplir sucesivamente tres funciones: o Con una actividad exonucleasa corta dos núcleótidos del extremo 3′ de cada una de las dos cadenas del ADN proviral. o Con una actividad endonucleasa (de doble cadena) corta el ADN del huésped en el punto de integración. No hay un lugar fijo en el genoma para que esto se realice, sino que ocurre en cualquier región muy accesible de la cromatina, lo que se supone que favorece la expresión del provirus, al coincidir esas regiones del genoma con las más transcritas.
Por último, con una actividad ligasa el ADN proviral es soldado, mediante sólo un enlace covalente en cada extremo, en el ADN celular.
En la actualidad existe un fármaco comercializado contra la actividad de la nucleasa, el ralteralvir.

La envoltura se basa en una bicapa lipídica, lo mismo que cualquier membrana biológica, y sus componentes estructurales básicos proceden de la membrana plasmática de la célula parasitada. Pero la envoltura porta además regularmente espaciadas 72 espículas, que son complejos proteicos integrados en la membrana formados por proteínas virales codificadas por el gen env. Cada espícula está formada por una pieza de la proteína gp41, integral en la membrana, y una cabeza externa formada por la proteína gp120, esencial para el acoplamiento con el exterior de ciertas células previo a su invasión. Entre los dos componentes de las espículas existe una unión no covalente. Las proteínas gp41 y gp120 se sintetizan como una sola poliproteína, gp160, con la información del gen env antes de que sea cortada por una proteasa de la célula. La proteína Env existe como trímero en la superficie de los viriones y las células infectadas.
Los fármacos inhibidores de la fusión funcionan contra la proteína gp41, para evitar su unión a los linfocitos.
Proteínas reguladoras.
Tat LLa proteína Tat existe en dos formas, una larga, de 101 restos aminoácidos de longitud, y otra más corta, de sólo 72. La segunda se produce cuando en fase temprana se produce una edición completa del ARNm viral, la primera cuando en una fase más tardía sólo se realiza una edición parcial. La proteína Tat (por transactivator) es imprescindible para la producción de nuevos viriones, que promueve activamente. La proteína se une a una región de 59 nucleótidos situada en el extremo 5′ del ARN viral llamada TAR (Transactivator Active Region) y actúa como un transactivador, algo excepcional, puesto que éstos suelen unirse al ADN, no al ARN. En cuanto este extremo inicial del genoma viral ha sido transcrito desde el ADN proviral, la proteína Tat se une a él y promueve su elongación favoreciendo la transcripción del resto de la cadena.
Rev
La proteína rev regula la expresión del ARN viral controlando el ritmo de exportación del ARNm.

Tat y Rev: acción conjunta
La acción sinergística de Tat y Rev fuertemente incrementa la expresión de proteínas virales. Los papeles que Tat y Rev desempeñan en la regulación transcripcional del VIH-1 y en la expresión de proteínas estructurales, respectivamente, hacen Tat y Rev esenciales para el ciclo de vida del VIH. Sus funciones facilitan la expresión de proteínas virales en dos etapas. Después de la integración del ADN proviral y de su transcripción en un nivel basal, solamente los RNAms de 2 KB se transportan al citoplasma. Esto permite la síntesis de Tat, Rev y de Nef. Tat y Rev entonces son transportadas al núcleo, donde actúan para aumentar la transcripción del ADN del provirus (Tat) y del transporte de todos los RNAms virales al citoplasma (Rev). La expresión de proteínas codificada por las clases de RNAm de 9 KB y 4 KB (Gag, Gag-Pol, Env, Vpr, Vif, y de Vpu) puede entonces ocurrir. Estudios donde se han mutado genes virales han determinado que Vif, Vpr, Vpu y Nef no son esenciales para la producción de partículas infecciosas en cultivos celulares «in-vitro». Sin embargo, la conservación de dichas proteínas accesorias en el genoma del VIH sugiere que todas desempeñan papeles importantes durante el ciclo infeccioso en el huésped. Los roles de estas proteínas serán descritos a continuación.
Proteínas accesorias
Vif: incremento en infectividad y protección del genoma viral
Vif es una proteína de 193 aminoácidos que está presente en bajos niveles adentro de los viriones, e interactúa con en RNA genómico viral. La división de esta proteína reduce la infectividad del VIH-1 en cultivos celulares y en modelos animales de patogénesis. No obstante, el mecanismo de acción de Vif se ha empezado a entender recientemente. La ausencia de Vif en partículas infecciosas no puede ser compensada con la expresión de Vif en las células infectadas. Estudios recientes han demostrado que Vif es requerida para eliminar la acción del factor ApoBEC3G, la cual es una deaminasa de citidinas, que convierte la citosina en uracilo, y emplea como sustrato el ADN de cadena sencilla. Además, esta enzima posiblemente actúa durante el ciclo de la transcriptasa inversa, modificando así la cadena negativa del DNA, porque esta es la fase en la cual el ADN de cadena sencilla está disponible. ApoBEC3G es selectivamente incorporada dentro de las partículas de VIH, resultando en un alto nivel de mutaciones en el genoma viral. Dado que estos altos niveles de mutación son perjudiciales para la viabilidad del virus, VIH ha evolucionado una estrategia para abolir esta poderosa barrera. Sin embargo, estudios recientes sugieren que ApoBEC3G no requiere su acción enzimática para tener efecto. Estudios más recientes han implicado que ApoBEC3G tiene un rol en la inhibición de ciertas fases en el ciclo de la transcriptasa inversa.

Vpu: facilita el desprendimiento de viriones en células infectadas
Vpu es una proteína de 81 aminoácidos que es insertada en membranas vía su terminal nitrogenado. Vpu se acumula en el aparato de Golgi y en endosomas celulares. Vpu es única en HIV-1 y no hay homólogos en lentivirus relacionados como el VIH-2 y el VIS. A Vpu se le han atribuido dos actividades.

Degradación de la proteína CD4
En la ausencia de Vpu, la proteína CD4 interactúa con la proteína viral gp160 recién sintetizada para formar un complejo insoluble, el cual retiene gp120 dentro de la celula. La región citoplásmica de Vpu se puede unir con CD4 y con la proteína β-TrCP. Esto induce la ubiquitinización de CD4 y su subsiguiente degradación por el proteasoma, incrementando así la expresión de gp120 en la superficie celular.
Realza en el desprendimiento de viriones de la membrana celular
Esta actividad depende de la región transmembranal de Vpu. En la ausencia de Vpu, los viriones se acumulan en la superficie celular en un estado parcialmente desprendido. Expresión de Vpu resulta en la liberación facilitada de viriones de la membrana celular. Remarcablemente, este efecto no está restringido solamente al VIH-1; Vpu también facilita el desprendimiento de otros virus no relacionados. El mecanismo por la cual esto ocurre es desconocido. Se ha sugerido que Vpu facilita la fluidez de la membrana celular por medio de un canal de cationes. También se ha sugerido que Vpu causa disrupción de interacciones entre proteínas del VIH y de la superficie celular; esto previene la endocitosis de viriones recientemente desprendidos de la célula…
Ciclo de vida
• Enlace y fusión: El VIH empieza su ciclo de vida cuando se liga a un receptor CD4 y a uno de dos correceptores en la superficie de un linfocito T CD4 +. Luego el virus se fusiona con la célula anfitriona. Después de la fusión, el virus libera el ARN, su material genético, dentro de la célula anfitriona. Además de linfocitos, el VIH puede llegar a las células dendríticas (células presentadoras de antígeno – CPA) gracias a la existencia de algunas lectinas como DC-SIGN y L-SIGN a las que se adhiere con alta afinidad, convirtiendo a las células dendríticas en importantes reservorios que ayudarán al VIH a infectar más linfocitos T CD4+.
• Transcripción inversa: Una enzima del VIH, conocida como transcriptasa inversa convierte la cadena simple del ARN vírico en cadena doble de ADN vírico.

• Integración: El nuevo ADN del VIH que se forma entra al núcleo de la célula anfitriona, donde una enzima del VIH llamada integrasa «esconde» el ADN vírico dentro del propio ADN de la célula anfitriona. El ADN del VIH integrado se llama provirus. El provirus puede permanecer inactivo por varios años sin producir nuevas copias del VIH o produciendo muy pocas.
• Transcripción: Cuando la célula anfitriona recibe señal para volverse activa, el provirus usa una enzima anfitriona llamada polimerasa del ARN para crear copias del material genómico del VIH y segmentos más cortos del ARN conocidos como ARN mensajero (ARNm). El ARNm se utiliza como modelo o patrón para la formación de cadenas largas de proteínas del VIH.
• Ensamblaje: La enzima del VIH llamada proteasa divide las cadenas largas de proteínas del VIH en pequeñas proteínas individuales. A medida que las proteínas pequeñas del VIH se unen a las copias del material genético del ARN del VIH, se ensambla una nueva partícula del virus.

• Gemación: El nuevo virus ensamblado «brota» de la célula anfitriona. Durante la gemación, el nuevo virus acapara parte de la envoltura exterior de la célula. A esta envoltura, que actúa como recubrimiento, le brotan combinaciones de proteína y azúcar, conocidas como glucoproteínas del VIH. Estas glucoproteínas del VIH son necesarias para que el virus se ligue al CD4 y a los correceptores. Las nuevas copias del VIH pueden ahora pasar a infectar a otras células.
Ciclo de replicación
Las células que el VIH invade son esencialmente los linfocitos T DC4+, pero también en menor medida los monocitos/macrófagos, las células dendríticas, las células de Langerhans y las células de microglía del cerebro. La replicación viral tiene pues lugar en tejidos diversos (de ganglios linfáticos, intestino, cerebro, timo,…). Los órganos linfoides, sobre todo los ganglios linfáticos, constituyen la principal sede de su replicación. El virus está presente en numerosos líquidos del organismo, en particular la sangre y las secreciones genitales.

La replicación del virus se desarrolla en las siguientes etapas:
• La fijación: representa la primera etapa en la invasión de una célula. Se basa en el reconocimiento mutuo y acoplamiento de proteínas de la envoltura del virión, las gp120 y gp41, y los receptores de la célula blanca, los CD4. Este reconocimiento no es posible sin ayuda de correceptores propios de las células susceptibles de ser invadidas; en el caso de los macrófagos son los CCR5 y en el caso de los LT4, los CXCR4, que interactúan con la proteína superficial. Macrófagos y LT4 tienen en común su principal receptor: el receptor CD4. Este reconocimiento es condición obligada para que el virus llegue a penetrar en la célula y continuar con el proceso de infección.
• La penetración: es el segundo paso, una vez reconocido el virión por los receptores de superficie, se vacía dentro de la célula fusionándose la envoltura lipídica del virión con la membrana plasmática de la célula. Protegidos por la cápside y las nucleocápsides, los dos ARN mensajeros que forman el genoma viral y sus proteínas asociadas se encuentran ahora en el citoplasma.luego ocurre la eliminación de las cubiertas proteicas, cápside y nucleocápsides, quedando el ARN vírico libre en el citoplasma y listo para ser procesado.

• La transcripción inversa del ARN vírico para formar ADNc (ADN complementario, monocatenario) con la misma información: Cada una de las dos moléculas de ARN llega desde el virión asociada a una molécula de transcriptasa inversa que se ocupa del proceso. Las dos moléculas de ADNc se asocian para formar una molécula de ADN, que es la forma química de guardar la información que una célula eucariota es capaz de procesar. Durante la preintegración el genoma viral es vulnerable a factores de transcripción como la TRIM5-α. Sin embargo, la cápside del VIH-1 no es reconocida por la forma humana del TRIM5-α por lo que no impide su integración.

• Integración del genoma vírico en el genoma de la célula huésped: Para ello penetra en el núcleo y se inserta en el ADN celular con ayuda de una integrasa, que procede del virión infectante.
• La transcripción del ADN vírico por los mecanismos normales de la célula: El resultado de la transcripción es un ARNm (ARN mensajero).El ARNm obtenido es complejo, constituido por una sucesión de intrones (partes no informativas) y exones (partes informativas). Debe ser procesado por cortes y reempalmes antes de que la información que contiene pueda servir para fabricar las proteínas correspondientes. Una vez procesado, el ARNm puede salir del núcleo a través de los poros nucleares.

La transcripción es llevado a cabo por la familia de factores de transcripción Rel/NF-kB. Sin embargo, este factor no existe en forma activa en los linfocitos T CD4 en estado de reposo celular y es inducido sólo en el curso de los procesos de activación inmunológica, lo que significa que la replicación del VIH depende sólo de la activación de los linfocitos T CD4 infectados.
• Traducción: Una vez en el citoplasma el ARNm proporciona la información para la traducción, es decir, la síntesis de proteínas, que es realizada a través del aparato molecular correspondiente, del que forman la parte fundamental los ribosomas. El resultado de la traducción no consiste inmediatamente en proteínas funcionales, sino en poliproteínas que aún deben ser cortadas en fragmentos.
Por acción de peptidasas específicas del VIH, las poliproteínas producto de la traducción son procesadas, cortándolas, para formar las proteínas constitutivas del virus.Las proteínas víricas fabricadas se ensamblan, junto con ARN provirales, para formar los componentes internos de la estructura del virión, los que constituyen la cápside y su contenido.

• Gemación: El último paso, ocurre cuando los nucleoides víricos se aproximan a la membrana plasmática y se hacen envolver en una verruga que termina por desprenderse, formando un nuevo virión o partícula infectante. En cada célula infectada se ensamblan varios miles de nuevos viriones, aunque muchos son incompletos y no pueden infectar.
Vías de transmisión del virus
El VIH sólo se puede transmitir a través del contacto entre fluidos corporales que poseen una alta concentración viral. El virus no se transmite de manera casual. De acuerdo con los CDC (Centros para el control y la prevención de enfermedades) de Estados Unidos, no se han encontrado casos en que abrazos, besos secos o saludos con las manos hayan sido causantes de infección. El virus ha sido aislado en la saliva, las lágrimas, la orina, el semen, el líquido preseminal, los fluidos vaginales, el líquido amniotico, la leche materna, el líquido cefalorraquideo y la sangre, entre otros fluidos corporales humanos.
Las tres formas de transmisión son:
• Sexual (relaciones sexuales sin protección). Por relaciones sexuales orales, vaginales o anales sin protección en el contacto de secreciones infectadas con la mucosa genital, rectal u oral de la otra persona. Por este motivo, se considera el VIH como una infección por trasmisión sexual
• Sanguínea (por sangre). Por contacto con sangre al compartir jeringas para la utilización de drogas inyectables u otros elementos para el consumo, así como otros elementos corto-punzantes como los usados durante la realización de piercings, tatuajes y escaricaciones. La transmisión también se puede producir por transfusiones de sangre o productos derivados de la sangre no controladas, lo que ha afectado particularmente a personas hemofílicas en los inicios de la epidemia. La transmisión sanguínea también puede producirse en personas trabajadoras de la salud que se exponen accidentalmente.
• Perinatal (de persona gestante a hijo). La transmisión puede ocurrir durante el embarazo, el parto o la lactancia. Actualmente es posible controlar la transmisión por esta vía para lo que un aspecto clave es el conocimiento del diagnóstico de la persona gestante para poder tomar las medidas necesarias tanto en la planificación previa al embarazo como desde el inicio del embarazo para la administración de tratamiento antirretroviral especialmente indicado para estas situaciones. En el momento del parto puede indicarse cesárea pero también es posible la realización de un parto vaginal. Se suprime la producción de leche, y con ello la lactancia, e incluso se da tratamiento antirretroviral al recién nacido.
Profilaxis post exposición
La profilaxis post-exposición (PEP) es el tratamiento antirretroviral a corto plazo para reducir la probabilidad de infección por el VIH después de haber sufrido una exposición potencial, ya sea ocupacionalmente o por otros motivos.
Debe proporcionarse como parte de las precauciones universales a la hora de reducir la exposición a fuentes de infección y siempre debería ser valorada por un equipo médico adecuadamente formado de un servicio de urgencias.
La administración de fármacos antirretrovirales como profilaxis post exposición debería abordarse con extrema cautela; estos medicamentos no pueden considerarse una alternativa a otras estrategias de prevención como el uso de condones o las medidas de bioseguridad.
Si se administra poco después de la exposición (dentro de las 72 horas), la PEP puede reducir el riesgo de infección por el VIH en más del 80 %. Completar el ciclo completo de 28 días de tratamiento con los antirretrovirales es fundamental para que la intervención sea efectiva.
En condiciones ideales, la profilaxis (tomar la medicación antirretroviral) debería iniciarse 1 o 2 horas después de la presunta exposición al VIH, pero nunca después de 72 horas. Los datos indican que cuanto antes se inicie el tratamiento, mayor es la probabilidad de éxito.
En algunos países, como en América del Norte y algunas zonas de Europa, la profilaxis post exposición puede obtenerse en los servicios de urgencias de cualquier hospital.29
Historia natural de la infección por VIH
La infección por VIH se presenta en diversas etapas, identificadas por un conjunto de síntomas e indicadores clínicos. En ausencia de un tratamiento adecuado, el virus se replica constantemente e infecta los linfocitos T- CD4, que constituyen una parte esencial del sistema inmunológico en los seres humanos. Por su parte, el sistema inmunológico del portador del VIH reacciona ante la presencia del virus y genera una respuesta que puede mantener la infección bajo control al menos por un tiempo, mediante la reposición de células defensivas. Al término de un período que se puede prolongar por varios años, el VIH se vuelve resistente a las defensas naturales del cuerpo y destruye el sistema inmune del portador. De esta manera, el individuo seropositivo queda expuesto a diversas enfermedades oportunistas y puede fallecer. El estadio de la enfermedad y su prognosis o el efecto de una terapia antiviral con antiretrovirales se mide bien con una combinación de dos parámetros:
• Población de linfocitos T CD4/ml. Se determina mediante citrometria del flujo
• Cuantificación de la carga viral (copias/ml), mediante PCR cuantitativa.
Fase aguda
La fase de la infección aguda por VIH inicia en el momento de la infección. El virus se propaga por el cuerpo de la persona infectada a través de sus fluidos corporales. En un plazo de días, el VIH infecta no sólo las células expuestas inicialmente (por ejemplo, las células de la mucosa vaginal o rectal en el caso de una infección por vía sexual) sino también los ganflios linfaaticos. Durante ese tiempo, el VIH se multiplica dentro del organismo hasta alcanzar niveles propios de la infección crónica. El tejido linfoide asociado a los intestinos constituye uno de los principales espacios del cuerpo humano donde tiene lugar la reproducción inicial del VIH por su alto porcentaje de linfocitos T CD4.
Un porcentaje importante de personas que contraen el virus no presenta síntomas de la infección en su fase aguda. Es decir, son pacientes asintomátiicos. Sin embargo, se calcula que entre el 40/50 %-90 % o hasta el 80 % de los casos de infección con VIH-1 presentan manifestaciones clínicas. El cuadro de la infección aguda es similar al de una mononucleosis infecciosa: fiebre, malestares musculares, inflamación de los ganglios, sudoración nocturna, diarrea, nauseas y vómito. La gran mayoría de los seropositivos no reciben diagnóstico del cuadro agudo de la infección por VIH, pues son síntomas compartidos por varias enfermedades. Por lo tanto, presentar un conjunto de síntomas como el descrito aquí no es indicador necesario de que una persona se haya infectado por VIH, aunque es recomendable que quien considere que ha estado expuesto a la transmisión y presente los síntomas, acuda a un especialista para recibir atención médica. El cuadro de la infección aguda por VIH aparece entre cinco y diez semanas después de la exposición al virus, y desaparece unos pocos días después.
El VIH ataca principalmente los linfocitos T CD4+, que forman parte del sistema inmune de los seres humanos. Aunque estas células por sí mismas no tienen una función de ataque contra células extrañas al cuerpo, tienen un papel importante en la respuesta inmunológica adaptativa. En una persona con buena salud, el número de linfocitos T CD4+ oscila entre 1200 y 500 μl. Durante la fase asintomática de la infección, la proporción de linfocitos infectados 1/1000-1/100 000, que aumentará progresivamente hasta llegar a 1/100 en la infección crónica. Durante la fase aguda de la infección, las pruebas tradicionales siempre darán negativo porque no detectan directamente el VIH, sino los anticuerpos producidos como respuesta por el sistema inmune, lo que ocurre alrededor de la 12a semana después de la exposición. En contraste, las pruebas de carga viral, que contabilizan el número de copias del ARN del virus en la sangre, arrojarán como resultado una elevada cantidad de copias del VIH durante la fase aguda de la infección
Fase crónica
La fase crónica de la infección por VIH se suele llamar también latencia clínica porque el portador es asintomático, es decir, no presenta síntomas que puedan asociarse con la infección. Esto no quiere decir que el virus se encuentre inactivo. Por el contrario, durante la fase crónica el VIH se multiplica incesantemente. Se calcula que, en un sujeto infectado, diariamente se producen entre mil y diez mil millones de nuevas partículas virales y son destruidos alrededor de cien millones de linfocitos T CD4. Los pacientes son asintomáticos gracias a que el sistema inmune tiene una gran capacidad para regenerar las células destruidas por el virus, pero pueden presentar adenopatías y la disminución del conteo de plaquetas en la sangre.
La reacción ante la presencia del virus termina por desgastar al sistema inmunológico. En ausencia de tratamiento, la mayoría de los portadores del virus desarrollan el síndrome de inmunodeficiencia adquirida (SIDA) en un plazo de 5 a 10 años. La causa de esto es que, mientras el virus sigue reproduciéndose de manera constante y aumenta la carga viral en su anfitrión, disminuye también la capacidad de recuperación del sistema inmune. Al término fase crónica, los pacientes desarrollan otras manifestaciones de la infección como dermatitis seborreica, úlceras bucales y foliculitis.

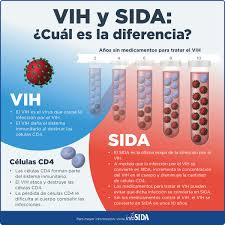
Síndrome de inmunodeficiencia adquirida
El sida constituye la etapa crítica de la infección por VIH. En esta fase de la infección, el portador del VIH posee un sistema inmunológico que probablemente sea incapaz de reponer los linfocitos T CD4+ que pierde bajo el ataque del VIH y también ha visto reducida su capacidad citotóxica hacia el virus. Este fenómeno coincide con el aumento en las tasas de replicación del virus, que merma la capacidad de reacción del anfitrión ante otros agentes causantes de enfermedades. De esta manera, el portador del virus es presa potencial de numerosas infecciones importunistas que le pueden conducir a la muerte. La neumonía por P jiroveci, el sarcoma de Karposi, la tuberculosis, la candidiasis y la infección por citomegalovirus son algunas de las infecciones más frecuentes que atacan a los seropositivos que han desarrollado sida.
La mayoría de los pacientes que han desarrollado sida no sobreviven más de tres años sin recibir tratamiento antirretroviral. Sin embargo, incluso en esta fase crítica el sida y el VIH pueden ser controlados mediante la terapia antirretroviral de gran actividad. Lo antirretrovirales brindan una mejor calidad de vida a un portador del VIH y aumentan sus posibilidades de supervivencia, tanto que hoy en día la enfermedad ha pasado de ser mortal a crónica.
La medicación con antirretrovirales no hace desaparecer el virus de los “reservorios” donde el VIH permanece de forma latente, pero sí consigue reducir la carga viral en sangre hasta el punto de ser indetectable (Carga Viral Indetectable (VIH)) e impedir su transmisión a otras personas.
La OMS (Organización Mundial de la Salud) ha clasificado a los infectados por VIH en 9 categorías en función del número de linfocitos T CD4 por mililitro de sangre y de la fase de la infección en la que se encuentren:
• Categoría A: pacientes en la fase precoz de la infección.
• Categoría B: pacientes en la fase crónica de la infección.
• Categoría C: pacientes en la fase final de la infección.
De este modo, sólo los infectados que se encuentren dentro de las categorías azules tienen SIDA, los demás pacientes sólo se consideran infectados por VIH.
Historia
Origen y evolución
Como otros agentes causantes de enfermedades infecciosas emergentes, el VIH pasó a los seres humanos por zoonosis, es decir por transmisión desde otras especies. La emergencia del sida y la identificación del VIH estimularon investigaciones que han permitido determinar que las variantes del VIH forman parte de un amplio grupo de lentivirus. El VIH es sumamente parecido a un virus que ataca a otros primates. Se trata del virus de la inmunodeficiencia de los simios (SIV), del que se conocen diversas cepas que se transmiten por vía sexual. A diferencia del VIH, el virus de los primates no causa inmunodeficiencia en los organismos que lo hospedan, salvo en el caso del salto de una especie a otra.
El VIH-1, responsable de la actual pandemia, ha resultado estar estrechamente relacionado con el SIVcpz, que infecta a poblaciones de la subespecie centroafricana del chimpancé común. El SIVcpz, a su vez, parece derivar por recombinación (un fenómeno que se produce fácilmente cuando infectan al mismo individuo dos cepas víricas diferentes) del SIVrcm, propio del mangabey de collar, y del SIVgsn, propio del avoem. Esta hipótesis es sostenida por el hecho de que tanto el VIH como las diversas cepas del SIV poseen el gen vpu además de que se han reportado transmisiones por SIV entre humanos en África ecuatorial. Las distribuciones actuales de las especies implicadas se solapan, y de los chimpancés se sabe que cazan monos pequeños para comerlos, lo que habría facilitado la coinfección por cepas diversas de SIV. La subespecie oriental del chimpancé, Pan troglodytes schweinfurthi, presenta también infección con una cepa propia del SIVcpz, pero genéticamente alejada del clado formado por el VIH-1 y las cepas de P.t.troglodytes. No se ha encontrado presencia del SIVcpz en la subespecie occidental, P. t. verus, aunque se observó la infección en cautividad de un individuo de esta subespecie.
El salto de la barrera de especie desde P. t. troglodytes a Homo sapiens sapiens se ha producido al menos tres veces, con variantes del VIH-1 que demuestran parentesco con distintas cepas, geográficamente más o menos localizadas, del SIVcpz. Así pues, el VIH-1 es un virus polifiletico. El grupo M del VIH-1, responsable de la pandemia actual, debió pasar a los seres humanos en la primera mitad del siglo XX. Los grupos O y N del VIH-1 están restringidos a África Occidental ecuatorial, con el grupo N presente sólo en Camerun. Con los datos actuales, parece claro que Pan troglodytes troglodytes es el reservorio desde el que se han producido repetidamente las infecciones humanas por los virus de cuya evolución procede el VIH-1
A su vez el VIH-2, extendido en África Occidental, procede del SIVsm, propio del mangavelle fuliginoso (Cercocebus atys atys), que habita las selvas costeras desde Senegal hasta Costa de Marfil. El análisis filogenético muestra que el paso a los seres humanos ha ocurrido también varias veces.
Los SIV identificados hasta ahora se encuentran, de forma específica y es en África donde parece tener su origen evolutivo este grupo monofilético de virus, genéticamente bien delimitado del resto de los lentivirus. La prevalencia (frecuencia de la infección) es variable entre especies y poblaciones, aunque no superior al 30 %, en las poblaciones afectadas de chimpancés, pero puede pasar del 50 % en poblaciones de otros primates, como Cercocebus ayds.
En todos los casos conocidos el virus parece encontrarse cerca del equilibrio con su huésped natural, como resultado probable de una más o menos larga coevolución, observándose generalmente sólo versiones muy atenuadas del síndrome de inmunodeficiencia, como una reducción limitada de linfocitos T CD4+, reducción que no compromete en general la vida del individuo, aunque en un ejemplar de Cercocebus atys se produjo un sida típico después de 18 años de incubación. Este dato hace pensar que, al menos en parte, es la baja longevidad, unida a una larga incubación, lo que hace que la inmunodeficiencia sobrevenida sea un resultado excepcional de la infección en monos.
Descubrimiento
Desde 1981 se detectaron casos sorprendentes de infección por Pneumocystis jiroveci (entonces designado Pneumocystis carinii), un hongo emparentado con las formas originales de los Ascomycetes, conocido por infectar a pacientes severamente inmunodeprimidos. Inicialmente se observó un grupo de casos semejantes en los que estaban implicados varones homosexuales y donde aparecían a la vez infección por citomegalovirus y candiasis. Se pensó primero que la causa debía estar ligada a prácticas comunes entre la población homosexual masculina.
Pronto empezaron a aparecer casos que afectaban a varones o mujeres heterosexuales usuarios de drogas intravenosas, así como a sus hijos; también entre pacientes no homosexuales ni bisexuales y con hábitos saludables que habían recibido transfusiones de sangre entera o de productos sanguíneos por su condición de hemofilicos. Pronto se pensó, por criterios básicamente epidemiológicos, que la causa debía ser un agente infeccioso que se transmitía de forma semejante a como lo hace el virus de la Hepatitis B.
Distintos equipos empezaron a buscar un virus asociado a los casos conocidos de inmunodeficiencia adquirida, tal vez un retrovirus como el que se sabía producía la inmunodeficiencia del gato o como el HTLV, productor de un tipo de leucemia. En 1983, en el Instituto Pasteur de París, un equipo dedicado a la investigación de la relación entre retrovirus y cáncer dirigido por J.C. Chermann, F. Barré-Sinoussi y L. Montagnier, encontró un candidato al que denominó lymphadenopathy-associated virus (virus asociado a la linfoadenopatía, LAV).
En 1984 el equipo de R. Gallo, descubridor del HTLV, único retrovirus humano conocido entonces, confirmó el descubrimiento, pero llamando al virus human T lymphotropic virus type III (virus linfotrópico T humano tipo III, con las siglas HTLV-III). Se produjo una subsecuente disputa sobre la prioridad en la que quedó claro que Gallo había descrito el virus sólo después de haber recibido muestras de los franceses. Como parte de la resolución del conflicto, el virus adquirió su denominación definitiva, human immunodeficiency virus (HIV) que en castellano se expresa como virus de la inmunodeficiencia humana (VIH).
En el mismo año, 1983, en que se identificó el virus, diversos equipos empezaron a trabajar en la secuencia de su genoma, publicada a principios de 1985, y comenzó también la caracterización de sus proteínas.
En 1985 se desarrolló técnica ELISA que permite conocer el alcance del virus y se descubrió un nuevo retrovirus en dos pacientes con SIDA, procedentes de Guinea Bissau y las Islas de Cabo Verde, diferente al VIH. Fue entonces cuando campo de estudio se traslada a África Occidental a investigar donde se llevó a cabo estudios en trabajadoras del sexo senegalesas con lo que se logró identificar un virus diferente al VIH inicial pero similar al descubierto en los dos pacientes de Guinea Bissau y las Islas de Cabo Verde, el VIH-2.
En 1987: se secuenció el genoma del VIH-2 y se confirmó que el VIH-2 era una zoonosis originario del VIS que diverge del VIH-1 en un 50% de su genoma y que en vez de tener el gen vpu (VIH-1), tenía el gen vpx (VIH-2).
Epidemiología
El VIH se ha convertido en una epidemia de dimensiones mundiales. El Programa Conjunto de Naciones Unidas sobre el VIH/sida (Onusida) coordina esfuerzos internacionales de científicos, gobiernos, iniciativa privada y organizaciones civiles dirigidos a actuar sobre la epidemia del VIH y sus efectos. Onusida observa el desarrollo epidemiológico de la infección por VIH en todo el mundo y emite un reporte sobre la situación de la epidemia cada dos años. Los informes de Onusida recopilan los datos provenientes de todos los países y dan una visión general de la evolución de la pandemia, sus efectos sociales, las estrategias adoptadas para controlarla.
Mundialmente, el modo más común de propagación del VIH sigue siendo la transmisión heterosexual. Entre 1981 y 2007, el sida había causado la muerte de aproximadamente 25 millones de personas alrededor de todo el mundo. En ese mismo año, 33 millones [30-36 millones] de personas estaban infectadas con VIH. La epidemia se ha estabilizado en cuanto que no ha aumentado la proporción de personas infectadas respecto a la población total. Además se ha observado una reducción del total mundial de nuevos casos de infección por VIH, de 3 millones [2,6-3,5 millones] en 2002 a 2,7 millones [2,2-3,2 millones] en 2007.
La región más afectada por la pandemia es África subsahariana, donde radican 21,5 millones [20,5-23,6 millones] de seropositivos. Esta cifra representa casi tres cuartos del total de casos calculados para todo el mundo. Esta región del mundo también presenta los índices más altos de mortalidad por sida y concentra el mayor número de nuevas infecciones.
VIH en adultos mayores
Históricamente se ha considerado al VIH como una enfermedad de adultos jóvenes; ahora se estima que aproximadamente el 25 % de los pacientes infectados con VIH en los Estados Unidos tienen 50 años de edad o más. Al principio de la epidemia del VIH, una proporción pequeña pero significativa de los adultos mayores fueron infectadas con el VIH a través de la transfusión sanguínea. Después esta tendencia cambió, los hombres mayores eran infectados a través de relaciones heterosexuales y en menor grado homosexuales o a través del uso de drogas intravenosas.
Estudios de investigación con pacientes VIH-positivos, en mayores de (50 años) frente a jóvenes de menos de (50 años) muestran que mientras más edad, los pacientes tienen más probabilidades de adquirir complicaciones médicas y más limitaciones en el funcionamiento físico.
La salud mental es también un factor importante que afecta a los pacientes. Tanto los hombres como las mujeres VIH-positivas han demostrado malestar psicológico asociado con las pruebas de infección por VIH, y la mayoría de los estudios han demostrado que las mujeres generalmente reportan síntomas psicopatológicos o complicaciones psicológicas mayores que en los hombres.
Uno de los problemas médicos para detectar el VIH y sus asociaciones en los adultos mayores son las respuestas poco fiables de esta población, que conducen a falsas suposiciones acerca del comportamiento de riesgo.
Fármacos contra el VIH
Existen numerosos fármacos dirigidos a evitar tanto la infección, como la progresión del ciclo vital del virus. Dichos fármacos se clasifican según la proteína a la que van dirigidos (esto es, el paso replicativo que inhiben en su uso). A continuación, se indican los diferentes fármacos existentes, así como el punto del ciclo replicativo que bloquean:
• Inhibidores de la unión o fusión: bloquean la penetración del virus en la célula diana inhibiendo la unión al correceptor CCR5 o CXCR4 de la superficie celular.
• Inhibidores de la transcriptasa inversa análogos de nucleósidos (ITIAN): impiden la replicación vírica mediante la inhibición de la síntesis del DNA complementario. Estos fármacos son reconocidos por el enzima como nucleósidos normales, de modo serán fosforilados e incorporados al DNA complementario.
• Inhibidores de la transcripatasa inversa no análogos de nucleósidos (ITINAN): inhiben la acción del enzima por otros mecanismos, evitando también la reproducción del virus.
• Inhibidores de la proteasa: bloquean la producción de viriones activos.
• Inhibidores de la integrasa: impiden que el virus introduzca su material genético en la célula diana.
En general, y dada la alta tasa de resistencias, está indicado el uso combinado de fármacos de diferentes grupos (politerapia), en lo que se viene llamando TARGA (Terapia AntirRetroviral de Gran Actividad).
El AZT por sí solo no puede destruir directamente el virus; lo que hace este fármaco es inhibir la enzima transcriptasa inversa, con lo que impide que el ARN del Virus se copie hacia ADNc bicatenario y, por consiguiente, evitar que se genere un provirus (el provirus es el ADNc que se integra al genoma de la célula huésped, en este caso es el linfocito T CD4+). Administrado de forma aislada, es decir, sin ser combinado con los otros medicamentos que componen el TARGA, puede incrementar las mutaciones en el virus que lo hagan más resistente y agresivo, anulando su eficacia terapéutica y acelerando el progreso de la enfermedad. Este riesgo disminuye notablemente cuando se combina con los otros medicamentos de la politerapia. También disminuye sensiblemente su toxicidad al reducirse y ajustarse con mejor precisión sus mínimas dosis efectivas en combinación con los otros componentes del TARGA.
Todas las mujeres embarazadas VIH+ deben tomar fármacos contra el virus, independientemente del número de linfocitos T CD4 y de la carga viral que presenten. El objetivo es reducir la carga viral hasta niveles indetectables y evitar, así, la transmisión vertical del virus. Además, cabe destacar que los tratamientos contra el VIH no aumentan el riesgo de que se produzcan defectos congénitos en el hijo/a.
Las mujeres embarazadas seropositivas pueden tomar los mismos medicamentos que las mujeres seropositivas no embarazadas, salvo en situaciones en las que el efecto secundario de algún fármaco comporte un riesgo elevado para la madre o el feto.
Las mujeres que tomaban un fármaco contra el virus antes del embarazo, pueden continuar tomándolo. Es importante que sigan correctamente el régimen de tratamiento (horas de toma, dosis…) y es posible que, una vez embarazadas, el médico decida cambiar algunos aspectos del régimen de tratamiento para reducir los efectos secundarios que se puedan producir.
El bebé es mucho más susceptible a infectarse por VIH durante el parto vaginal al pasar por el canal del parto, ya que está expuesto a la sangre y otros fluidos de la madre. Por eso, en esta situación se administra zidovudina vía intravenosa en madres con carga viral alta ( >1000 copias/mL) o con carga viral desconocida. Este fármaco puede atravesar la placenta y proporcionar la protección necesaria al bebé para que no pueda ser infectado por el VIH.
Además, para estas mujeres embarazadas con carga viral alta o desconocida, también se recomienda una cesárea electiva o programada para reducir el riesgo de transmisión vertical del VIH. En estos casos, el parto se programa para la semana 38 del embarazo, 2 semanas antes de la fecha estimada.
Detección del VIH
Debido a que no existe ninguna manifestación clínica característica de la infección de VIH, la prueba para detectar la infección ha de llevarse a cabo mediante pruebas de diagnóstico molecular en un laboratorio. Aunque desde 2002 la FDA(Administración de Alimentos y Medicamentos por sus siglas en inglés) aprobó el uso de pruebas rápidas para uso por personal capacitado que brinda un resultado en 20 minutos aproximadamente, que se usan fuera de laboratorio, que funciona como una Prueba inmunocromatográfica Cualitativa para la detección de Anticuerpos para los Tipos de Virus de la innmunodeficiencia humana 1 y 2 (HIV-1 y HIV-2), así como HIV-1 Tipo 0, en suero o plasma humano. Cada dispositivo de prueba contiene una banda de prueba que consta de una almohadilla de prueba, una almohadilla dorada impregnada con un conjugado de proteína HIV y oro coloidal, una tira de nitrocelulosa con proteínas recombinantes VIH inmovilizadas como línea de Prueba y un reactivo vinculante de anticuerpos como línea de Control, un material absorbente para facilitar el flujo a través del dispositivo, dicha prueba se aplica ya sea en saliva, como en sangre (se toma de igual manera que la glucosa en la yema de algún dedo) y se entrega un resultado (Reactivo/No reactivo). Para el caso que el resultado sea reactivo, será necesario entonces aplicar la prueba de laboratorio para descartar un falso positivo. El Dispositivo de Prueba Rápida de HIV 1&2 es un ensayo de tamizado. Puesto que la producción de anticuerpos al VIH puede retrasarse después de la exposición inicial, la no reactividad con esta prueba no debe ser considerado evidencia concluyente hasta confirmarse el diagnóstico de igual manera un resultado negativo no descarta la posibilidad de exposición a VIH o infección con el VIH antes de esta prueba solo se conocía la prueba más habitual para detectar la presencia de VIH es la prueba de inmunodetección denominada ELISA. Con esta técnica se pretende detectar los anticuerpos específicos que el organismo produce como respuesta a la presencia del virus. Cabe destacar que, en países donde la prevalencia de la enfermedad es baja, ante un resultado positivo mediante un ELISA, no se debe informar al paciente de la presencia de VIH sin haber confirmado antes la prueba mediante un western blog. Sin embargo, en países o determinados grupos sociales donde el VIH presenta una alta prevalencia, no será necesaria la confirmación con western blot.
Por lo tanto, en la mayoría de los casos la seropositividad frente al VIH se detecta a partir de una extracción sanguínea del sujeto con la que se realizará la determinación de anticuerpos anti-VIH por alguna técnica de cribado como la ya nombrada ELISA u otras parecidas. La prueba diagnóstica dirigida al VIH tiene una especificidad del 99 % y una sensibilidad del 99 %.
Aparte de las pruebas conocidas como pruebas de anticuerpos (ELISA), existen otros tipos de pruebas:
• Pruebas de combinación permiten detectar anticuerpos y antígenos del VIH en la sangre.Estas pruebas permiten detectar la infección por VIH antes que una prueba de anticuerpos contra el mismo. Los antígenos ya anticuerpos en sangre tardan en ser detectables de 2 a 6 semanas.Las pruebas de combinación son cada vez más comunes en los Estados Unidos.
• Las pruebas de ácido nucleico examinan la presencia del VIH en la sangre. Permiten detectar el VIH entre 7 y 28 días después de contagiarse. Este tipo de pruebas es muy caro y no se usa habitualmente a menos que la persona haya tenido una exposición de alto riesgo o una posible exposición y presente los primeros síntomas de infección por el VIH. Entre este tipo de pruebas se encuentra la PCR-nested o anidada (amplificación de un amplicón contenido dentro de otro producto de una amplificación previa), que posee muy alta especificidad y sensibilidad pero no cuantifica. Para detectar el virus insertado en el genoma, el ADN proviral, se utiliza una PCR anidada. Para detectar el ARN viral, se usa RT-PCR anidada.
MVA-B: Perspectivas actuales de investigación
En la actualidad, un grupo de investigación español del CSIC, ha presentado una posible vacuna contra el VIH, la vacuna MVA-B.
La vacuna MVA-B se denomina así por su composición a partir del virus Vaccinia Modificado de Ankara (MVA), y la letra B procede del subtipo de VIH contra el que lucha, el más prevalente en Europa.
Esta vacuna se encuentra en la fase I de desarrollo y ha presentado una alta seguridad y eficacia. Un 90 % de los voluntarios vacunados con MVA-B han generado una respuesta inmunitaria defensiva contra el VIH, y el 85 % de ellos la ha mantenido, al menos durante un año.
Para el desarrollo de la vacuna MVA-B, se han introducido cuatro genes del VIH (Gag, Pol, Nef y Env) en la secuencia genética de vaccinia. Si el sistema inmune está sano reacciona frente al MVA, y los genes de VIH insertados en su secuencia no son capaces de infectar a los humanos, lo que garantiza la seguridad del ensayo clínico. Pese a estos resultados esperanzadores, la vacuna todavía no puede ser comercializada, ya que ha de concluir con éxito todas las fases de desarrollo del ensayo clínico para poder salir al mercado.
ENFERMEDAD DE ALZHEIMER.

La enfermedad de Alzheimer (EA), denominada demencia senil de tipo Alzheimer (DSTA) o simplemente alzhéimer, es una enfermedad neurodegenerativa que se manifiesta como deterioro cognitivo y trastornos conductuales. Se caracteriza en su forma típica por una pérdida de la memoria inmediata y de otras capacidades mentales (tales como las capacidades cognitivas superiores), a medida que mueren las células nerviosas (neuronas) y se atrofian diferentes zonas del cerebro. La enfermedad suele tener una duración media aproximada —después del diagnóstico— de 10 años, aunque esto puede variar en proporción directa con la severidad de la enfermedad al momento del diagnóstico. L
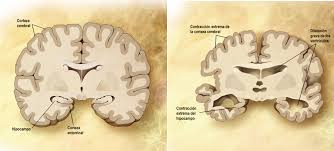
La enfermedad de Alzheimer es la forma más común de demencia, es incurable y terminal, y aparece con mayor frecuencia en personas mayores de 65 años de edad, aunque también en raros casos puede desarrollarse a partir de los 40 años.
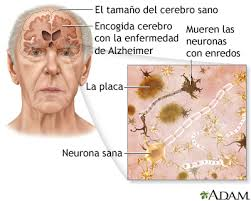
Muchas personas experimentan olvidos o retrasos leves de memoria, que son parte del proceso normal de envejecimiento. Todos tenemos dificultad ocasional para recordar una palabra o el nombre de alguien. Sin embargo, una persona con la enfermedad de Alzheimer u otros tipos de demencia, encontrará estos síntomas cada vez más frecuentes y graves.

Los signos que indican la enfermedad de Alzheimer pueden incluir:
• Cambios en la personalidad
• Deterioro en la capacidad de movimiento o al caminar
• Dificultad para comunicarse
• Bajo nivel de energía
• Pérdida de memoria
• Cambios de estado de ánimo
• Problemas de atención y orientación
• Incapacidad de resolver operaciones aritméticas sencillas.

Los sintomas son como una entidad nosológica definida fueron identificados por el psiquiatra alemán Emil Kraepelin mientras que la neuropatología característica fue observada por primera vez por el psiquiatra y neurólogo alemán Alois Alzheimer en 1906. Así pues, el descubrimiento de la enfermedad fue obra de ambos psiquiatras, que trabajaban en el mismo laboratorio. Sin embargo, dada la gran importancia que Kraepelin daba a encontrar la base neuropatológica de los desórdenes psiquiátricos, decidió nombrar a la enfermedad Alzheimer en honor a su compañero.

Por lo general, el síntoma inicial es la inhabilidad de adquirir nuevos recuerdos, pero suele confundirse con actitudes relacionadas con la vejez o el estrés. Ante la sospecha de alzhéimer, el diagnóstico se realiza con evaluaciones de conductas cognitivas, así como neuroimágenes, si están disponibles. A medida que progresa la enfermedad, aparecen confusión mental, irritabilidad y agresión, cambios del humor, trastornos del lenguaje, pérdida de la memoria de corto plazo y una predisposición a aislarse a medida que declinan los sentidos del paciente. Gradualmente se pierden las funciones biológicas, que finalmente conllevan a la muerte. El pronóstico para cada individuo es difícil de determinar. El promedio general es de siete años, menos del 3% de los pacientes viven más de 14 años después del diagnóstico.

La causa de la enfermedad de Alzheimer permanece desconocida, aunque las últimas investigaciones parecen indicar que están implicados procesos de tipo priónico. Las investigaciones suelen asociar la enfermedad a la aparición de placas seniles y ovillos neurofibrilares. Los tratamientos actuales ofrecen moderados beneficios sintomáticos, pero no hay tratamiento que retrase o detenga el progreso de la enfermedad. No obstante, casos preliminares de asociación de demencia por Alzheimer con la enfermedad celiatica mostraron la mejoría con el seguimiento de una dieta sin gluten.

Para la prevención del alzhéimer, se han sugerido varios hábitos conductuales, pero no hay evidencias publicadas que destaquen los beneficios de esas recomendaciones, incluyendo la estimulación mental y la dieta equilibrada. El papel que juega el cuidador del sujeto con alzhéimer es fundamental, aun cuando las presiones y la demanda física de esos cuidados pueden llegar a ser una gran carga personal.

El Día Internacional del Alzheimer se conmemora el 21 de diciembre, fecha elegida por la OMS y la Federación Internacional de Alzheimer, en la cual se llevan a cabo actividades en diversos países para concienciar y ayudar a prevenir la enfermedad. La Organización Mundial de la Salud (OMS) realizó en 2015 su Primera Conferencia Ministerial de la OMS sobre la Acción Mundial contra la Demencia.
Historia.
Médicos griegos y romanos asociaron la vejez con la demencia. Pero no fue hasta 1901 cuando el psiquiatra alemán Alois Alzheimer identificó el primer caso de lo que se conoce hoy como enfermedad de Alzheimer en una mujer de 51 años de edad; esta mujer se llamaba Auguste Deter. El investigador hizo seguimiento de su paciente hasta su muerte en 1906, y entonces pudo observar el cerebro. Después de este momento pudo reportar públicamente por primera vez el caso.

Tras la muerte de la mujer, Alzheimer le examinó el cerebro con un microscopio. Anotó las alteraciones de las «neurofibrillas», elementos del citoesqueleto teñidos con una solución de plata.
Durante los siguientes cinco años, la literatura médica reportó al menos once casos similares, algunos de ellos utilizando ya el término «enfermedad de Alzheimer».La enfermedad fue categorizada por primera vez por Emil Kraepelin después de la supresión de algunos elementos clínicos concomitantes como delirios y alucinaciones, así como características histológicas irrelevantes para la enfermedad, como los cambios arterioscleróticos, los cuales figuraban en el informe original sobre Auguste D. En la octava edición de su libro de texto de Psiquiatría, publicado en 1910, incluyó a la enfermedad de Alzheimer, denominada también por Kraepelin «demencia presenil», como un subtipo de demencia senil.

Durante la mayor parte del siglo XX, el diagnóstico del alzhéimer era reservado para las personas entre las edades de 45 y 65 años con síntomas de demencia. La terminología ha cambiado desde 1977, cuando, en una conferencia sobre alzhéimer, se llegó a la conclusión de que las manifestaciones clínicas y patológicas de la demencia presenil y senil eran casi idénticas, aunque los autores también agregaron que ello no descartaba la posibilidad de que tuviesen causas diferentes. Esto, a la larga, conllevó a que se hiciera el diagnóstico del alzhéimer independientemente de la edad. El término demencia senil del tipo Alzheimer fue empleado durante un tiempo para describir el trastorno en aquellos mayores de 65 años, mientras que la enfermedad clásica de Alzheimer se reservaba para los de edades menores. Finalmente, el término enfermedad de Alzheimer fue aprobado oficialmente en la nomenclatura médica para describir a individuos de todas las edades con un patrón de síntomas: característica, curso de la enfermedad y neuropatología comunes.
Epidemiología
La incidencia en estudios de cohortes muestra tasas entre 10 y 15 nuevos casos cada mil personas al año para la aparición de cualquier forma de demencia y entre 5 a 8 para la aparición del alzhéimer. Es decir, la mitad de todos los casos nuevos de demencia cada año son pacientes con alzhéimer. También hay diferencias de incidencia dependiendo del sexo, ya que se aprecia un riesgo mayor de padecer la enfermedad en las mujeres, en particular entre la población mayor de 85 años.
La prevalencia es el porcentaje de una población dada con una enfermedad. La edad avanzada es el principal factor de riesgo para sufrir alzhéimer: mayor frecuencia a mayor edad. En los Estados Unidos, la prevalencia del alzhéimer fue de un 1,6 % en el año 2000, tanto en la población general como en la comprendida entre los 65 y 74 años. Se apreció un aumento del 19 % en el grupo de los 75-84 años y del 42 % en el mayor de 84 años de edad,sin embargo, las tasas de prevalencia en las regiones menos desarrolladas del mundo son inferiores. La Organización Mundial de la Salud estimó que en 2005 el 0,379 % de las personas a nivel mundial tenían demencia y que la prevalencia aumentaría a un 0,441 % en 2015 y a un 0,556 % en 2030. Por otro lado, para el año 2010 la Alzheimer’s Disease International ha estimado una prevalencia de demencia del 4,7 % a nivel mundial para personas con 60 años o más, representando por cierto cifras al alza respecto a varios estudios publicados con anterioridad (10 % superiores a las estimadas para The Lancet en 2005). Otro estudio estimó que en el año 2006, un 0,4 % de la población mundial (entre 0,17–0,89 %; valor absoluto aproximadamente 26,6 millones con un rango entre 11,4–59,4 millones) se vio afectada por alzhéimer y que la prevalencia triplicaría para el año 2050.

En 2015 la Primera Conferencia de la OMS sobre la Acción Mundial contra la Demencia estimó en 47.5 millones el número de casos en el mundo.
Etiología.
Las causas del alzhéimer no han sido descubiertas completamente. Existen tres principales hipótesis para explicar el fenómeno: el déficit de la acetilcolina, la acumulación de amiloide o tau y los trastornos metabólicos.
Hipótesis colinérgica. La más antigua de ellas, y en la que se basan la mayoría de los tratamientos disponibles en el presente, es la hipótesis colinérgica, la cual sugiere que el alzhéimer se debe a una reducción en la síntesis del neurotrasmisor acetilcolina . Esta hipótesis no ha recibido un apoyo global por la razón de que los medicamentos que tratan una deficiencia colinérgica tienen reducida efectividad en la prevención o cura del alzhéimer, aunque se ha propuesto que los efectos de la acetilcolina dan inicio a una acumulación a tan grandes escalas que conlleva a la neuroinflamación generalizada que deja de ser tratable simplemente promoviendo la síntesis del neurotransmisor.
Hipótesis de los trastornos metabólicos. Algunas investigaciones recientes han relacionado la demencia, incluyendo la enfermedad de Alzheimer, con desórdenes metabóliicos particularmente con la hiperglucemía y la resistencia a la insulina . La expresión de receptores de la insulina ha sido demostrada en las neuronas del sistema nervioso central, preferentemente en las del hipocampo . En estas neuronas, cuando la insulina se une a su receptor celular, se promueve la activación de cascadas de señalización intracelular que conducen al cambio de la expresión de los genes relacionados con los procesos de plasticidad sináptica y de las enzimas relacionadas con el despeje de la misma insulina y del beta-amiloide. Estas enzimas degradantes de insulina promueven la disminución de la toxicidad debida al amiloide en modelos animales.

En esencia, el alzhéimer puede ser considerado como una forma de diabetes de cerebro que tiene elementos tanto de resistencia a la insulina como de deficiencia de insulina. Para consolidar este concepto, se ha propuesto que el alzhéimer sea conocido como «diabetes tipo 3». El alzhéimer está asociado con una progresiva resistencia a la insulina cerebral en ausencia de DM2, de resistencia a la insulina periférica o de obesidad. Estudios post mortem demostraron que tanto molecular como bioquímicamente, así como la transducción de señales anormales en el alzhéimer eran virtualmente idénticas a lo que ocurría en la diabetes mellitus tipo I y II. Al administrar localmente drogas prodiabéticas, como la estreptozotocina, estas generan daño cognitivo, con deficiencia espacial y de memoria, así como neurodegeneración típica de alzhéimer, pero no generan diabetes mellitus.

Exposición al aluminio. Ha existido polémica en torno al papel que tiene el aluminio como factor de riesgo para la enfermedad de Alzheimer, y esta hipótesis ha sido abandonada por numerosos científicos en 2014. Algunos estudios han mostrado en 2016, que el aluminio se asocia a varios procesos neurofisiológicos que provocan la característica degeneración del alzhéimer. Sin embargo, algunas personas que han estado crónicamente expuestas al aluminio, a través de alimentos o agua, no han mostrado ningún síntoma de la enfermedad. La probable explicación es que su intestino mantiene la función de barrera protectora, que evita el paso de sustancias tóxicas a la sangre y las consecuentes reacciones.
Consumo de gluten. Se han documentado casos de asociación de demencia por Alzheimer con el consumo de gluten y la mejoría con el seguimiento de una dieta sin gluten.
Hipótesis de las proteínas β-amiloide y tau.
Otra hipótesis propuesta en 1991 se ha relacionado con el acumulo anómalo de la proteína beta-amiloide (también llamada amiloide Aβ) y tau en el cerebro de los pacientes con alzhéimer. En una minoría de pacientes, la enfermedad se produce por la aparición de mutaciones en los genes, PSEN1 y PSEN2 y en el gen de la APP, localizado en el cromosoma 21. En este último caso la enfermedad aparece clásicamente en personas con el Síndrome de Down (trisomía en el cromosoma 21), casi universalmente en los 40 años de vida y se transmite de padres a hijos (por lo que existen, habitualmente, antecedentes familiares de alzhéimer en los pacientes que desarrollan la enfermedad en edades precoces). Esa relación en el cromosoma 21, y la tan elevada frecuencia de aparición de la enfermedad en las trisomías de ese cromosoma, hacen que la teoría sea muy evidente.

Para poder hablar de las presenilinas, debemos recordar que el alzhéimer está caracterizado por depósitos amiloideos en el cerebro (como apoya la segunda teoría de este artículo). Su componente principal es el péptido beta-amiloide de 42 aminoácidos (βA42), en cuyo proceso de producción es fundamental la participación de la γ-secretasa, la cual depende a su vez de las presenilinas (PSEN).

De esta manera se instituye un nuevo grupo de moléculas implicadas en la génesis de la enfermedad del Alzheimer y fundamental para su comprensión. Además son de notable actualidad debido a que su descubrimiento es relativamente reciente y a las posibilidades que ofrecen como dianas terapéuticas. Para comprender qué son estas moléculas, debemos señalar que se trata de un grupo de sustancias peptídicas producidas principalmente en el cerebro. Hay dos tipos, PSEN 1 y PSEN 2, con una estructura similar. La función principal que desempeñan ambas PSEN consiste en el procesamiento proteolítico de numerosas proteínas de membrana de tipo 1, entre ellas la APP, formando parte de la γ secretasa; de ahí la importancia de las PSEN en la enfermedad de Alzheimer, ya que a través de la regulación de la γ secretasa determinan la forma de Aβ que se genera y por tanto su acumulación en el tejido cerebral. El alzhéimer de inicio temprano se ha relacionado con mutaciones en el cromosoma 21, que contiene el gen de la PPA, y los cromosomas 14 y 1, que codifican para PSEN1 y PSEN2, respectivamente. Estas mutaciones tienen como resultado, entre otros efectos, el aumento de la concentración de βA, mientras que el alzhéimer de inicio tardío se relaciona con mutaciones en el gen de la apolipoproteina E. El gen que codifica la PSEN1, del que se conocen 177 mutaciones distintas, es el responsable de la aparición del alzhéimer de inicio tan temprano como a los 23 años. La mutación de la PSEN2 es la causante de menos del 1% de los casos de alzhéimer autosómicos dominantes, influyendo más en estos portadores los factores ambientales. La revisión de diferentes artículos nos ha conducido a la conclusión de que existe una relación entre las PSEN y el alzhéimer. Actualmente el avance en las técnicas de secuenciación del genoma ha aportado gran cantidad de información sobre las PSEN, su función, la localización en nuestros genes, la implicación de estas en la formación de βA, etc. No obstante, hoy en día aún se observan lagunas en cuanto a los mecanismos moleculares en los que están implicadas las PSEN.

Otro gran factor de riesgo genético es la presencia del gen de la APOE4(apolipoproteína relacionada con la hiperlipoproteinemia y la hipercolesterolemia familiar), el cual tiende a producir una acumulación amiloide en el cerebro antes de que aparezcan los primeros síntomas del alzhéimer. Por ende, la deposición del amiloide Aβ tiende a preceder la clínica del alzhéimer. Otras evidencias parten de los hallazgos en ratones genéticamente modificados, los cuales solo expresan un gen humano mutado, el de la APP, el cual invariablemente les causa el desarrollo de placas amiloides fibrilares. Se descubrió una vacuna experimental que causaba la eliminación de estas placas pero no tenía efecto sobre la demencia.

Los depósitos de las placas no tienen correlación con la pérdida neuronal. Esta observación apoya la hipótesis tau, la cual defiende que es esta proteína la que da inicio a la cascada de trastornos de la enfermedad de Alzheimer. De acuerdo con este modelo, las tau hiperfosforiladas adoptan formas anómalas, distribuyéndose en largas hileras. Eventualmente forman ovillos de neurofibrillas dentro de los cuerpos de las células nerviosas. Cuando esto ocurre, los microtubulos se desintegran, colapsando el sistema de transporte de la neurona. Ello puede dar inicio a las primeras disfunciones en la comunicación bioquímica entre una neurona y la otra y conllevar la muerte de estas células.

Según un trabajo realizado por un equipo de científicos de la Universidad de Cambridge, en Reino Unido cuyos resultados se publican en Brain, la proteína tau, causante de la muerte de las células nerviosas, se disemina por todo el cerebro en la enfermedad de Alzheimer y, por lo tanto, bloquear su propagación puede evitar que la enfermedad aflore.

Se considera que los síntomas del Alzheimer están causados por la acumulación en el cerebro de dos proteínas anómalas: la proteína beta amiloide y la proteína tau. Se cree que la beta amiloide se produce primero, fomentando la aparición y la diseminación de tau, y que es esta última la que destruye las células nerviosas, mermando la memoria y las funciones cognitivas.
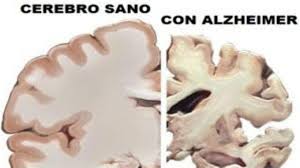
Hasta hace unos años solo era posible observar la acumulación de estas proteínas mediante el examen de los cerebros de los pacientes post mortem. Sin embargo, los recientes desarrollos en la tomografía por emisión de positrones (PET) han permitido a los científicos comenzar a visualizar su acumulación en pacientes que están vivos.

La forma en que tau se presenta en todo el cerebro ha sido tema de especulación entre los científicos y se han propuesto tres hipótesis:
1. Una hipótesis es que la tau anómala comienza en un punto y desde ahí se propaga a otras regiones, desencadenando una reacción en cadena. Esta idea es conocida como la «propagación transneuronal» y está respaldada por estudios en ratones. Cuando a un ratón se le inyecta una proteína tau humana anómala, esta se propaga rápidamente por todo el cerebro. Sin embargo, debe reconocerse que la cantidad de tau inyectada es mucho más alta que los niveles de tau observados en cerebros humanos y la proteína se propaga rápidamente por el cerebro de un ratón mientras se disemina lentamente por el cerebro humano.

2. La hipótesis de la «vulnerabilidad metabólica», que considera que la proteína tau se produce localmente en las células nerviosas, pero que algunas regiones presentan mayores demandas metabólicas y, por lo tanto, son más vulnerables a la proteína. En estos casos, tau es un marcador de sufrimiento en las células.
3. Y la última hipótesis, del «soporte trófico», también plantea que algunas regiones cerebrales son más vulnerables que otras, pero que esto tiene menos que ver con la demanda metabólica y más con la falta de nutrición en la región o con los patrones de expresión génica.
Los avances en el escaneo PET permiten actualmente hacer comparaciones entre estas hipótesis.
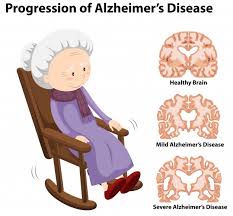
Los científicos analizaron las conexiones funcionales dentro de los cerebros de los pacientes de Alzheimer y lo compararon con los niveles de tau. Sus hallazgos respaldan la idea de la propagación transneuronal, es decir, que tau comienza en un lugar y se propaga. Al mismo tiempo encontraron que no se cumplían las predicciones para las últimas dos hipótesis.

Según Thomas Cope, el primer autor del estudio, la propagación transneuronal es la correcta, las áreas del cerebro que están más conectadas tienen la mayor acumulación de la proteína tau anómala y la transmitirán a sus conexiones. En la enfermedad de Alzheimer, la región cerebral más común para tau que aparece primero es la región de la memoria, en el área de la corteza entorrinal, que está al lado del hipocampo. Los primeros síntomas en el Alzheimer tienden a ser problemas de memoria y el estudio de Cope sugiere que la tau luego se propaga por el cerebro, infectando y destruyendo las células nerviosas a medida que avanza, lo que hace que los síntomas del paciente empeoren progresivamente.

Esta confirmación es importante porque indica que se puede ralentizar o incluso detener la progresión de la enfermedad de Alzheimer mediante el desarrollo de fármacos para evitar que tau se mueva a lo largo de las neuronas.
Hipótesis microbiológicas.
Existen numerosas hipótesis (método científico) que han sido manejadas a lo largo de la historia de la patogenia del Alzheimer, que no han sido confirmadas por estudios independientes posteriores. En 2013, un equipo de investigación publicó un estudio estadístico en el que halló correlación entre las infecciones fúngicas diseminadas y la enfermedad de Alzheimer. Esta hipótesis no han sido confirmada. En 2018 la hipótesis de un equipo, plantea que la bacteria Porphyromonas gingivalis podría ser una de las causas de enfermedad de Alzheimer. Esta hipótesis no han sido confirmada.
Patogenia
La enfermedad de Alzheimer se caracteriza por la pérdida de neuronas y sinapsis en la corteza cerebral en ciertas regiones subcorticales. Esta pérdida resulta en una atrofia de las regiones afectadas, incluyendo una degeneracion en el lobulo y parietal y partes de la corteza frontal y la circunvolución cingulada

Neuropatología.
Las placas son depósitos densos, insolubles, de la proteína beta-amiloide y de material celular que se localizan fuera y alrededor de las neuronas. Estas continúan creciendo hasta formar fibras entretejidas dentro de la célula nerviosa, los llamados ovillos. Es probable que muchos individuos, en su vejez, desarrollen estas placas y ovillos como parte del proceso normal de envejecimiento. Sin embargo, los pacientes con alzhéimer tienen un mayor número en lugares específicos del cerebro, como el lóbulo temporal.

Bioquímica
La enfermedad de Alzheimer se ha definido como una enfermedad que desdobla proteínas o proteopatía, debido a la acumulación de proteínas Aβ y tau, anormalmente dobladas, en el cerebro. Las placas neuríticas están constituidas por pequeños peptidos de 39–43 aminoacidos de longitud, llamados beta-amiloides (abreviados A-beta o Aβ). El beta-amiloide es un fragmento que proviene de una proteína de mayor tamaño conocida como Proteína Precursora de Amiloide (APP, por sus siglas en ingles). Esta proteína es indispensable para el crecimiento de las neuronas, para su supervivencia y su reparación postdaño. En la enfermedad de Alzheimer, un proceso aún desconocido es el responsable de que la APP sea dividida en varios fragmentos de menor tamaño por enzimas que catalizan un proceso de proteolisis. Uno de estos fragmentos es la fibra del beta-amiloide, el cual se agrupa y deposita fuera de las neuronas en formaciones microscópicamente densas conocidas como placas seniles.
La enfermedad de Alzheimer se considera, debido a la agregación anormal de la proteína tau , como una tauropatía. Las neuronas sanas están compuestas por citoesqueleto, una estructura intracelular de soporte, parcialmente hechas de microtúbulos. Estos microtúbulos actúan como rieles que guían los nutrientes y otras moléculas desde el cuerpo neuronal hasta los extremos de los axones y viceversa. Cada proteína tau estabiliza los microtúbulos cuando es fosforilada y por esa asociación se le denomina proteína asociada al microtúbulo. En el alzhéimer, la tau debido a cambios químicos que resultan en su hiperfosforilación, se une con otras hebras tau creando ovillos de neurofibrillas y de esta manera, desintegra el sistema de transporte de la neurona.

Patología
No se ha explicado por completo cómo la producción y agregación de los péptidos Aβ (beta Amiloides) desempeñan un papel rol en el alzhéimer. La fórmula tradicional de la hipótesis amiloide apunta a la acumulación de los péptidos Aβ como el evento principal que conlleva la degeneración neuronal. La acumulación de las fibras amiloides, que parece ser la forma anómala de la proteína responsable de la perturbación de la homeostasis del ión calcio intracelular, induce la muerte celular programada, llamada apoptosis. Se sabe también que la Aβ se acumula selectivamente en las mitocondrias de las células cerebrales afectadas en el alzhéimer y que es capaz de inhibir ciertas funciones enzimáticas, así como alterar la utilización de la glucosa por las neuronas.

Varios mecanismos inflamatorios y la intervención de las citoquinas pueden también desempeñar un papel en la patología de la enfermedad de Alzheimer. La inflamación es el marcador general de daño en los tejidos en cualquier enfermedad y puede ser secundaria al daño producido por el alzhéimer, o bien, la expresión de una respuesta inmunológica.

Genética.
La gran mayoría de los pacientes de esta enfermedad, tienen o han tenido algún familiar con alzhéimer. También hay que decir que en una pequeña proporción de los pacientes, el Alzheimer es debido a una generación autosómica dominante, que hace que la enfermedad aparezca de forma temprana. En menos de un 10% de los casos, el alzhéimer aparece antes de los 60 años de edad como consecuencia de mutaciones autosómicas dominantes, representando, apenas, un 0,01% de todos los casos. Estas mutaciones se han descubierto en tres genes distintos: el gen de la proteína precursora de amiloide (la APP) y los genes de las preselinias 1 y 2. Si bien la forma de aparición temprana de la enfermedad de Alzheimer ocurre por mutaciones en tres genes básicos, la forma más común no se ha podido explicar con un modelo puramente genético. La presencia del gen de la apolipoproteína E es el factor de riesgo genético más importante para padecer Alzheimer, pero no permite explicar todos los casos de la enfermedad.
En 1987, se descubrió la relación de la enfermedad de Alzheimer con el cromosoma 21. Esto fue importante porque la mayoría de los afectados por el «síndrome de Down», o trisomía del cromosoma 21, padecen lesiones neuropatológicas similares a las del Alzheimer. Dentro del cromosoma 21 encontramos el gen PPA. John Hardy y sus colaboradores en 1991 afirmaron que este gen estaba implicado en la Enfermedad de Alzheimer en un reducido número de familias. Sin embargo, se considera que de entre 5-10% de los familiares con la enfermedad precoz la padecen debido a una mutación de este gen. Las investigaciones dentro de este gen se han centrado en el péptido Ab (todas las mutaciones se encuentran alrededor de este péptido). Las mutaciones producían un aumento de las concentraciones del péptido Ab. Esto llevó a la formación de la hipótesis de «cascada amieloide» en los años 90. La «cascada amieloide» consiste en que la gran producción de Ab llevaría a la formación de depósitos en formas de placas seniles. Estas placas seniles serían nocivas para las células que producirían ovillos neurofibrilares, la muerte celular y la demencia. Más tarde se vio en un grupo amplio de familias el ligamiento de la enfermedad del Alzheimer con el cromosoma 14. Pero esto llevó a una cadena de errores y con ello unas conclusiones erróneas. Rudy Tanzi y Peter St George-Hyslop en 1995, mediante las técnicas de clonaje descubrieron otro gen S182 o Presenilin-1 (PS1). Este gen se encuentra entre los dominios 9 y 8 de transmembrana (con dos regiones hidrofílicas) y se le han encontrado más de 30 mutaciones. Este gen interviene en procesos de apoptosis y es fundamental durante el desarrollo. La mayoría de las mutaciones del gen Presenilin-1 (PS1) provocan un cambio en la estructura primaria. La PS1 y la enfermedad del Alzheimer no tienen una clara relación, pero hay que destacar que los pacientes tuvieron mutaciones que aumentan Ab en el plasma. Poco más tarde se descubrió un nuevo gen que se denomina presenilina-2 (PS2) y también provoca el ascenso en la concentración de Ab, aunque las mutaciones observadas son de menor cantidad que los otros genes (PPA y PS1). La PS2 está formada por 8-9 dominios transmembrana.

La mayoría de las mutaciones en el gen de la APP y en los de las presenilinas, aumentan la producción de una pequeña proteína llamada beta-amiloide (abeta 2), la cual es el principal componente de las placas seniles.

Aunque la mayoría de los casos de Alzheimer no se deben a una herencia familiar, ciertos genes actúan como factores de riesgo. Un ejemplo es la transmisión familiar del alelo e4 del gen de la apolipoproteína E. Este gen se considera un factor de riesgo para la aparición de Alzheimer esporádico en fases tardías, produciendo un 50% de los casos Alzheimer. Además de este, alrededor de 400 genes han sido también investigados por su relación con el Alzheimer esporádico en fase tardía. Así pues, los genetistas coinciden en que hay más genes que actúan como factores de riesgo, aunque también afirman que existen otros que tienen ciertos efectos protectores que conllevan a retrasar la edad de la aparición del Alzheimer. Un ejemplo es la alteración en el gen de la reelina, que contribuye a aumentar el riesgo de aparición del alzhéimer en mujeres.
Cuadro clínico
Predemencia.
Los primeros síntomas se confunden, con frecuencia, con la vejez o estres en el paciente. Una evaluación neuropsicológica detallada es capaz de revelar leves dificultades cognitivas hasta 8 años antes de que la persona cumpla los criterios de diagnóstico. Estos signos precoces pueden tener un efecto sobre las actividades de la vida diaria. La deficiencia más notable es la pérdida de memoria, manifestada como la dificultad de recordar hechos recientemente aprendidos y una inhabilidad para adquirir nueva información. Dificultades leves en las funciones ejecutivas-atención , planificación, flexibilidad y razonamiento abstracto— o trastornos en la memoria semántica—el recordar el significado de las cosas y la interrelación entre los conceptos— pueden también ser síntomas en las fases iniciales del alzhéimer. Puede aparecer apatía, siendo uno de los síntomas neuropsiquiátricos persistentes a lo largo de la enfermedad. La fase preclínica de la enfermedad es denominada por algunos deterioro cognitivo leve, pero aún existe debate sobre si el término corresponde a una entidad diagnóstica independiente o si, efectivamente, es la primera etapa de la enfermedad.
Demencia inicial.
La disminución en la destreza de la coordinación muscular de pequeños movimientos, como el tejer, comienzan a aparecer en el paciente de Alzheimer en las fases iniciales de la enfermedad.
Los síntomas en esta fase inicial van desde una simple e insignificante, pero a veces recurrente, pérdida de memoria (como la dificultad en orientarse uno mismo en lugares como calles al estar conduciendo el automóvil), hasta una constante y más persuasiva pérdida de la memoria conocida como memoria a corto plazo, presentando dificultades al interactuar en áreas de índole familiar como el vecindario donde el individuo habita.
Además de la recurrente pérdida de la memoria, una pequeña porción de los pacientes presenta dificultades para el lenguaje, el reconocimiento de las percepciones —agnosia— o en la ejecución de movimientos —apraxia— con mayor prominencia que los trastornos de la memoria. El alzhéimer no afecta las capacidades de la memoria de la misma forma. La memoria a largo plazo o memorias episódicas, así como la memoria semántica o de los hechos aprendidos y la memoria implicita, que es la memoria del cuerpo sobre cómo realizar las acciones (tales como sostener el tenedor para comer), se afectan en menor grado que las capacidades para aprender nuevos hechos o el crear nuevos recuerdos.
Los problemas del lenguaje se caracterizan, principalmente, por reducción del vocabulario y disminución en la fluidez de las palabras, lo que conlleva a un empobrecimiento general del lenguaje hablado y escrito. El paciente con alzhéimer suele ser capaz de comunicar adecuadamente las ideas básicas. También aparece torpeza al realizar tareas motoras finas, tales como escribir, dibujar o vestirse, así como ciertas dificultades de coordinación y de planificación. El paciente mantiene su autonomía y solo necesita supervisión cuando se trata de tareas complejas. En esta etapa es frecuente que la persona se desoriente en la calle y llegue a perderse, por lo que se recomienda tomar precauciones:
• Colocándole en la muñeca una pulsera con un número de teléfono de contacto.
• Avisando a los que conocen la situación, para que alerten a la familia en caso de encontrar deambulando al enfermo de alzhéimer.
• Usando un localizador GPS para personas con alzhéimer, con el que la familia siempre pueda saber dónde está. Existen localizadores por teleasistencia, en los que el cuidador debe de llamar a una teleoperadora para saber la ubicación del enfermo que lleva el dispositivo, y localizadores directos, en los que el cuidador tiene un receptor con el que, pulsando un botón, ve en la pantalla un mapa y la posición exacta de la persona enferma.

Demencia moderada.
Conforme avanza la enfermedad, los pacientes pueden realizar tareas con cierta independencia (como usar el baño), pero requerirán asistencia para tareas más complejas (p. ej. ir al banco, pagar cuentas, etc…). Paulatinamente llega la pérdida de aptitudes, como las de reconocer objetos y personas. Además, pueden manifestarse cambios de conducta como, por ejemplo, arranques violentos incluso en personas que jamás han presentado este tipo de comportamiento.
Los problemas del lenguaje son cada vez más evidentes debido a una inhabilidad para recordar el vocabulario, lo que produce frecuentes sustituciones de palabras erróneas, una condición llamada parafasia. Las capacidades para leer y escribir empeoran progresivamente. Las secuencias motoras complejas se vuelven menos coordinadas, reduciendo la habilidad de la persona para hacer sus actividades rutinarias. Durante esta fase, también empeoran los trastornos de la memoria y el paciente empieza a dejar de reconocer a sus familiares y seres más cercanos. La memoria a largo plazo, que hasta ese momento permanecía intacta, se deteriora.

El paciente con alzhéimer no muere por la enfermedad, sino por infecciones secundarias, como una llaga de presión o úlcera de decúbito, lesiones que se producen cuando una persona permanece en una sola posición por mucho tiempo.
En esta etapa se vuelven más notorios los cambios en la conducta. Las manifestaciones neuropsiquiátricas más comunes son las distracciones, el desvarío y los episodios de confusión al final del día (agravados por la fatiga, la poca luz o la oscuridad), así como la irritabilidad y la labilidad emocional, que incluyen llantos o risas inapropiadas, no premeditada e incluso resistencia a las personas a cargo de sus cuidados. En el 30% aproximadamente de los pacientes aparecen ilusiones en el reconocimiento de personas. También puede aparecer la incontingencia urinaria Estos síntomas estresan a los familiares y a las personas al cuidado del paciente y pueden verse reducidos si se le traslada a un centro de cuidados a largo plazo.

Demencia avanzada.
La enfermedad trae deterioro de la masa muscular, perdiéndose la movilidad, lo que lleva al enfermo a un estado de encamamiento, la incapacidad de alimentarse a sí mismo, junto a la incontinencia, en aquellos casos en que la muerte no haya llegado aún por causas externas (infecciones por úlceras o neumonia, por ejemplo). El lenguaje se torna severamente desorganizado, llegándose a perder completamente. A pesar de ello, se conserva la capacidad de recibir y enviar señales emocionales. Los pacientes no podrán realizar ni las tareas más sencillas por sí mismos y requerirán constante supervisión, quedando así completamente dependientes. Puede aún estar presente cierta agresividad, aunque es más frecuente ver extrema apatía y agotamiento.

Síntomas y etapas
Los principales síntomas del Alzheimer son perdidas de memoria a corto plazo, (desconocimiento de la enfermedad), confabulaciones para tapar los huecos en el recuerdo de corto plazo (mienten o inventan cosas para rellenar el vacío o laguna en sus relatos), estado depresivo, problemas de atención, problemas con la orientación, cambios de personalidad, dificultades con el lenguaje y cambios de humor inexplicables, desorientación, entre otras. Esta enfermedad tiene tres etapas en las cuales presenta diferentes síntomas.
Etapa 1 o leve. Cuando la enfermedad comienza a manifestarse, las personas con Alzheimer tienden a ser menos enérgicas y espontáneas. Muestran pérdidas mínimas de la memoria y cambios de humor, y tardan en aprender y reaccionar. También se hacen aislados, evitan la gente que no conocen y los lugares nuevos y prefieren lo familiar. Los individuos se confunden, tienen dificultades para la organización y planificación, se pierden fácilmente y ejercen un pobre juicio. Pueden tener dificultad para realizar las tareas rutinarias, y tienen dificultad para comunicarse y comprender material escrito. Si la persona está empleada, la pérdida de memoria puede comenzar a afectar el rendimiento en el trabajo. Todo esto les causa frustración y enojo.
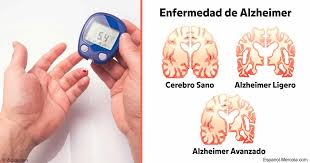
Etapa 2 o moderada. En esta etapa, la persona con la enfermedad de Alzheimer todavía puede realizar tareas simples independientemente, pero puede necesitar ayuda con actividades complicadas. Los enfermos olvidan los acontecimientos recientes y su historia personal, y cada vez están más desorientados y desconectados de la realidad. Pueden confundir su pasado con el presente, y les cuesta comprender la situación actual, fecha y hora. También pueden tener problemas para reconocer personas familiares. Aumentan los problemas con el habla y la comprensión, la lectura y la escritura son más difíciles, y el individuo tiende a inventar palabras. Los afectados ya no pueden estar seguros solos y pueden deambular. Mientras los pacientes de la enfermedad de Alzheimer se hacen más conscientes de esta pérdida de control, se pueden volver depresivos, irritables e inquietos o apáticos y aislados. Pueden experimentar trastornos del sueño y tienen dificultad para comer, vestirse y asearse.

Etapa 3 o grave. Durante esta fase final, la gente puede perder la capacidad para alimentarse a sí misma, hablar, reconocer personas y el control de las funciones corporales. Su falta de memoria se agrava y puede llegar a ser casi completa. La atención constante es necesaria. En un estado físico debilitado, el paciente puede llegar a ser vulnerable a otras enfermedades y problemas respiratorios, sobre todo cuando tiene que estar confinado a la cama.
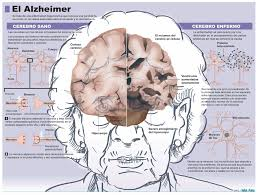
Diagnóstico.
El diagnóstico se basa primero en la historia y la observación clínicas, del profesional de la salud y la que es referida por los familiares, basada en las características neurológicas y psicológicas, así como en la ausencia de condiciones alternativas: un diagnóstico de exclusión.
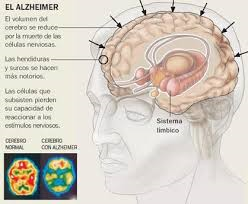
Luego durante unas semanas o meses se realizan pruebas de memoria y de funcionamiento o evaluación intelectual. También se efectúan análisis de sangre y escaner para descartar diagnósticos alternativos. No existe un test pre mortem para diagnosticar concluyentemente el alzhéimer. Se ha conseguido aproximar la certeza del diagnóstico a un 85%, pero el definitivo debe hacerse con pruebas histológicas sobre tejido cerebral, generalmente obtenidas en la autopsia (McKhann 1984) . Las pruebas de imagen cerebral – Tomografia Axial Computarizada(TAC), Resonancia Magnetica (RMN), Tomografía por emisión de Positrones(TEP) o la tomografía computarizada por emisión de protón único— pueden mostrar diferentes signos de que existe una demencia, pero no especifican de qué demencia se trata. Por tanto, el diagnóstico de la enfermedad de Alzheimer se basa tanto en la presencia de ciertas características neurológicas y neuropsicológicas, como en la ausencia de un diagnóstico alternativo y se apoya en el escáner cerebral para detectar signos de demencia. Actualmente se están desarrollando nuevas técnicas de diagnóstico basadas en el procesamiento de señales electroencefalográficas.

Una vez identificada, la expectativa promedio de vida de los pacientes que viven con la enfermedad de Alzheimer es aproximadamente de 7 a 10 años, aunque se conocen casos en los que se llega antes a la etapa terminal, entre 4 y 5 años; también existe el otro extremo, donde pueden sobrevivir hasta 21 años.
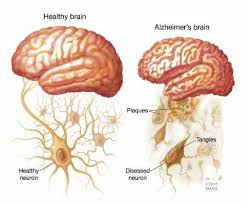
Criterios de diagnóstico.
La Asociación del Alzheimer es el organismo que ha establecido los criterios diagnósticos más comúnmente usados, registrados en los Criterios NINCDS-ADRDA del Alzheimer. Estas pautas requieren que la presencia de un trastorno cognitivo y la sospecha de un síndrome demencial sean confirmadas con una evaluación neuropsicológica con vistas a categorizar el diagnóstico de Alzheimer en dos: posible o probable. La confirmación histológica, que incluye un examen microscópico del tejido cerebral, se precisa para el diagnóstico definitivo del Alzheimer. Estos criterios incluyen que la presencia de un trastorno cognitivo y la sospecha de un síndrome demencial sean confirmados por evaluaciones neuropsicológicas para distinguir entre un diagnóstico posible o uno probable de la enfermedad de Alzheimer. Se ha mostrado fiabilidad y validez estadística entre los criterios diagnósticos y la confirmación histológica definitiva. Son ocho los dominios cognitivos que con más frecuencia se dañan en el alzhéimer: la memoria, el lenguaje, la percepción, la atención, las habilidades constructivas y de orientación, la resolución de problemas y las capacidades funcionales. Estos parámetros son equivalentes a los evaluados en los Criterios NINCDS-ADRDA publicados por la Asociación Americana de Psiquiatría.
Herramientas de diagnóstico
Las evaluaciones neuropsicológicas pueden ayudar al diagnóstico del alzhéimer. En ellas se acostumbra hacer que el paciente copie dibujos similares a la imagen, que recuerde palabras, lea o sume.
Las evaluaciones neuropsicológicas, inclusive el examen minimental, son ampliamente usadas para evaluar los trastornos cognitivos necesarios para el diagnóstico de EA. Otra serie de exámenes más comprensivos son necesarios para una mayor fiabilidad en los resultados, especialmente en las fases iniciales de la enfermedad. El examen neurológico en los inicios del alzhéimer es crucial para el diagnóstico diferencial del alzhéimer y otras enfermedades.
Las entrevistas a familiares también sirven para evaluar la enfermedad. Los cuidadores pueden proveer información y detalles importantes sobre las habilidades rutinarias, así como la disminución en el tiempo de la función mental del paciente. El punto de vista de la persona a cargo del paciente es de especial importancia debido a que el paciente, por lo general, no está al tanto de sus propias deficiencias. Muchas veces, los familiares tienen desafíos en la detección de los síntomas y signos iniciales de la demencia y puede que no comuniquen la información de manera acertada al profesional de salud especializado.
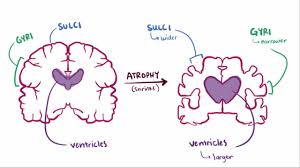
Los exámenes adicionales pueden proporcionar información de algunos elementos de la enfermedad y tienden a ser usados para descartar otros diagnósticos. Los exámenes de sangre pueden identificar otras causas de demencia que no sea el alzhéimer, que pueden ser, en pocos casos, enfermedades reversibles. El examen psicológico para la depresión sería de valor, puesto que la depresión puede aparecer de manera concomitante con el alzhéimer, o bien ser la causa de los trastornos cognitivos.
En los casos en que estén disponibles imágenes neurológicas especializadas, como la TEP o la tomografía de fotón único pueden servir para confirmar el diagnóstico del alzhéimer junto con las evaluaciones del estatus mental del individuo. La capacidad de una tomografía computarizada por emisión de fotón único, para distinguir entre el alzhéimer y otras posibles causas en alguien que ya fue diagnosticado de demencia, parece ser superior a los intentos de diagnóstico mediante exámenes mentales y la historia del paciente.
Una nueva técnica, conocida como PiB PET, se ha desarrollado para tomar imágenes, directamente y de forma clara, de los depósitos beta-amiloides in vivo, con el uso de un radiofármaco que se une selectivamente a los depósitos Aβ. Otro marcado objetivo reciente de la enfermedad de Alzheimer es el análisis del liquido cefalorraquideo en busca de amiloides beta o proteínas tau. Ambos avances de la imagen médica han derivado en propuestas para cambiar los criterios diagnósticos.
Tratamiento Actualmente la enfermedad de Alzheimer es incurable y terminal.
El tratamiento del Alzheimer se sustenta en dos pilares complementarios: el tratamiento no farmacológico y el tratamiento farmacológico. Las intervenciones psicosociales se usan conjuntamente con el tratamiento farmacológico.
Tratamientos farmacológicos
Se ha probado cierta eficacia de parte de los fármacos anticolinesterados, estos presentan una acción inhibidora de la colinesterasa, la enzima encargada de descomponer la acetilcolina (neurotransmisor que falta en la enfermedad de Alzheimer y que incide sustancialmente en la memoria y otras funciones cognitivas). Se han incorporado al tratamiento de la enfermedad nuevos fármacos que intervienen en la regulación de la neurotransmisión glutaminergica. Con todo esto se ha mejorado un poco el comportamiento del enfermo en cuanto a la apatía, la iniciativa, la capacidad funcional y las alucinaciones, mejorando su calidad de vida. Sin embargo, es preciso remarcar que desde 2008 los registros de la mejoría obtenida con dichos fármacos es discreta, es decir, no se ha conseguido alterar el curso de la demencia subyacente.
El primer fármaco anticolinesterásico comercializado fue la tacrina, que hoy ha dejado de emplearse por su hepatotoxicidad. En 2008, en Europa y Norteamerica existían 4 fármacos disponibles, tres de ellos son inhibidores de la acetilcolinesterasa: donepezilo (comercializado como Aricept), rivastigmina (comercializado como Exelon o Prometax) incluyendo el parche de Exelon, y galantamina (comercializado como Reminyl). Los tres presentan un perfil de eficacia similar con parecidos efectos secundarios. Estos últimos suelen ser alteraciones gastrointestinales, anorexia y trastornos del ritmo cardíaco. El cuarto medicamento es un antagonista de los receptores de la MMDA, la memantina. Ninguno de los cuatro se indica para retardar o detener el progreso de la enfermedad.
La reducción en la actividad de las neuronas colinérgicas es una de las características reconocidas de la enfermedad de Alzheimer. Los inhibidores de la acetilcolinesterasa se emplean para reducir la tasa de degradación de la acetilcolina, manteniendo así concentraciones adecuadas del neurotransmisor en el cerebro y deteniendo su pérdida causada por la muerte de las neuronas colinérgicas. Existen evidencias de que estos medicamentos tienen eficacia en las etapas leves y moderadas de la enfermedad, aunque un poco menos de que sean útiles en la fase avanzada. Solo el donepezilo se ha aprobado para este estado de la demencia. El uso de estos fármacos en los trastornos cognitivos leves no ha mostrado ser capaz de retardar la aparición del alzhéimer. Los efectos adversos más comunes incluyen nauseas y vomitos , ambos ligados al exceso colinérgico que de ellos deriva. Estos efectos aparecen entre el 10 y el 20 % aproximadamente de los tratados y tienen severidad leve a moderada. Entre los efectos secundarios menos frecuentes figuran calambres musculares, disminución de la función cardiaca, disminución del apetito y del peso corporal y un incremento en la producción de jugo gastrico.
La memantina es un fármaco con un mecanismo de acción diferente, que está indicado en las fases moderadas y avanzadas de la enfermedad. Su mecanismo de acción teórico se basa en antagonizar los receptores NMDA glutaminérgicos, usado en un principio como un agente antigripal. El glutamato es un neurotrasmisor excitatorio del sistema nervioso central. Al parecer, un exceso de estimulación glutaminérgica podría producir o inducir una serie de reacciones intraneuronales de carácter tóxico, causando la muerte celular por un proceso llamado excitotoxicidad, que consiste en una sobreestimulación de los receptores del glutamato. Esta excitotoxicidad no solo ocurre en pacientes con alzhéimer, sino también en otras enfermedades neurodegenerativas, como la Enfermedad de Parkinson y la esclerosis multiple. Los ensayos clínicos han demostrado una eficacia moderada en estos pacientes y un perfil de efectos secundarios aceptable. En 2005 se aprobó también su indicación en fases moderadas de la enfermedad, pero los efectos en las fases iniciales son aún desconocidos. Los efectos adversos de la memantina son infrecuentes y leves e incluyen alucinaciones, confusión, dolor de cabeza y fatiga. La combinación de memantina y donepezilo ha mostrado ser estadísticamente significativa pero marginalmente exitosa desde el punto de vista clínico.
Además existen fármacos que mejoran algunos de los síntomas que produce esta enfermedad, entre los que se encuentran ansiolíticos, hipnóticos, neurolépticos y antidepresivos. Los fármacos antipsicóticos se indican para reducir la agresión y la psicosis en pacientes con alzhéimer que tienen problemas de conducta, pero se usan con moderación y no de forma rutinaria por razón de los serios efectos secundarios, como eventos cerebrovasculares, trastornos extrapiramidales y una reducción cognitiva.
Tratamientos no farmacológicos.
Retrasar el avance del alzhéimer
El avance de la enfermedad puede ser más rápido o más lento en función del entorno de la persona con alzhéimer. No es una situación fácil y la familia tendrá que hacer grandes esfuerzos para ofrecerle a la persona con alzhéimer un entorno lo más favorable posible. Aceleradores de la enfermedad. Se consideran aceleradores las siguientes situaciones: estrés familiar, cambios bruscos en las rutinas diarias, cambio a un domicilio nuevo y desconocido (como son las residencias de mayores).
Retardadores de la enfermedad.
Se consideran retardadores las situaciones nombradas a continuación: ambiente familiar feliz, hacer ejercicio, socializar con los amigos u otras personas.
Intervención psicosocial
Existen ciertas evidencias de que la estimulación de las capacidades cognitivas ayuda a ralentizar la pérdida de estas funciones y habilidades. Esta estimulación consiste en trabajar aquellas áreas que aún conserva el paciente, de forma que el entrenamiento permita compensar las pérdidas que el paciente está sufriendo con la enfermedad.
Las intervenciones psicosociales se usan conjuntamente con el tratamiento farmacológico y se clasifican en abordajes orientados al comportamiento, las emociones, lo cognitivo y la estimulación. Las investigaciones sobre la efectividad de estas intervenciones aún no se encuentran disponibles y, de hecho, rara vez son específicas al alzhéimer, enfocándose en la demencia en general.
Las intervenciones en el área del comportamiento intentan identificar y reducir los antecedentes y consecuencias de los problemas de la conducta. Este abordaje no ha mostrado éxito en mejorar el funcionamiento general del paciente, en especial en relación con su entorno, pero ha podido ayudar a reducir ciertos problemas específicos del comportamiento, como la incontinencia urinaria. Siguen faltando datos de calidad sobre la efectividad de estas técnicas en otros problemas como las deambulaciones del paciente.
Las intervenciones orientadas a las emociones comprenden la terapia de validación, la terapia de reminiscencia, la psicoterapia de apoyo, la integración sensorial (también denominada snoezelen) y la terapia de presencia estimuladora. La psicoterapia de apoyo ha tenido poco estudio científico formal, pero algunos especialistas la encuentran de utilidad en pacientes con trastornos leves. La terapia de reminiscencia incluye la discusión de experiencias del pasado de manera individual o en grupo, muchas veces con la ayuda de fotografías, objetos del hogar, música y grabaciones u otras pertenencias del pasado. En esta terapia, igualmente, no hay muchos estudios de calidad sobre su efectividad, aunque puede resultar beneficiosa para la restauración cognitiva y el humor. El tratamiento con presencias estimuladas se basa en las teorías de la adherencia e implica escuchar voces grabadas de los familiares y seres más cercanos del paciente con alzhéimer. Las evidencias preliminares indican que dichas actividades reducen la ansiedad y los comportamientos desafiantes.
Finalmente, la terapia de validación se basa en la aceptación de la realidad y la experiencia personal de otras personas, mientras que la integración sensorial se basa en ejercicios guiados que estimulan los sentidos. Aún no hay suficientes evidencias que apoyen el uso de estas terapias en pacientes con alzhéimer.
La finalidad de las terapias cognitivo-conductuales, que incluyen la orientación y la rehabilitación cognitiva, es reducir las distorsiones cognitivas. La orientación hacia la realidad consiste en presentar información acerca de la época, el lugar o la persona, con el fin de aliviar su entendimiento acerca de sus alrededores y el lugar que ellos desempeñan en dichos sitios. Por otro lado, el entrenamiento cognitivo intenta mejorar las capacidades debilitadas al ejercitar las habilidades mentales del paciente. Ambos ejercicios han mostrado cierta efectividad en el mejoramiento de las capacidades cognitivas. Sin embargo, en algunos estudios, estos efectos fueron transitorios y en otros tenían un efecto negativo, pues añadían frustración al paciente, según los reportes.
Los tratamientos orientados a la estimulación incluyen la arteterapia, la musicoterapia y las terapias asistidas por máscotas, el ejercicio físico y cualquier actividad recreaccional. La estimulación tiene apoyo modesto al ser aplicada con la intención de mejorar la conducta, el humor y, en menor grado, el funcionamiento del paciente. Si bien son efectos importantes, el principal beneficio reportado entre las terapias de estimulación es el mejoramiento en las rutinas de la vida diaria del paciente.
Cuidados
Debido a que el alzhéimer no tiene cura, con el tiempo el paciente cae en un estado de imposibilidad de autosuficiencia para cuidar de sí mismo, por lo que los cuidados por terceros son una medida vital para esa deficiencia y deben ser abordados cuidadosamente durante el curso de la enfermedad.
En las fases tempranas y moderadas, las modificaciones al ambiente donde vive el paciente y a su estilo de vida, pueden darle seguridad y reducir las cargas al cuidador. Algunos ejemplos de dichas modificaciones son la adherencia a rutinas simplificadas, como son la colocación de candados, el uso de una pulsera con el número de teléfono del cuidador (o soluciones más avanzadas como un localizador por GPS), el etiquetado de los objetos del hogar y el uso de utensilios modificados para la vida diaria. Puede llegar el punto en que el paciente no sea capaz de alimentarse a sí mismo, de modo que debe empezar a ingerir sus alimentos en porciones más pequeñas o en dietas no sólidas con la ayuda de otras personas. Cuando aparezca una dificultad para tragar, puede que sea indicado el uso de sondas gástricas. En tales casos, la efectividad médica y ética de tener que continuar alimentando al paciente son consideraciones importantes que deben tomar los cuidadores y los familiares del individuo. Las restricciones físicas rara vez están indicadas en cualquier fase de la enfermedad, aunque hay situaciones en que son necesarias para prevenir que el paciente con alzhéimer se dañe a sí mismo o a terceros.
A medida que progresa la enfermedad, pueden aparecer distintas manifestaciones médicas, como, las enfermedades orales y dentales, úlceras de presión, desnutricion, problemas de higiene o infecciones respiratorias, urinarias, dermatológicas u oculares, entre otras. El manejo cuidadoso del paciente puede prevenir dichos problemas, pero si llegan a producirse, deben ser tratados bajo supervisión médica. Durante las etapas finales de la enfermedad, el tratamiento se centra en mantener la calidad de vida hasta el fallecimiento.
Tratamientos en fase de investigación
Vacunas.
Igualmente se están realizando experimentos con vacunas, basados en la idea de que si el sistema inmune puede ser entrenado para reconocer y atacar la placa beta-amiloide, podría revertirse la deposición de amiloide y parar la enfermedad. Los resultados iniciales en animales fueron prometedores. Sin embargo, cuando las primeras vacunas se probaron en seres humanos en 2002, se produjo inflamación cerebral, (meningoencefalitis), en una pequeña proporción de los participantes en el estudio, por lo que se detuvieron las pruebas. Se continuó estudiando a los participantes y se observó una mejora en la lentitud del progreso de la enfermedad. Recientemente se ha descubierto que la inflamación cerebral estaba producida por una serie de péptidos que se incluían con la vacuna AN-179, por lo que se está investigando la creación de una vacuna que no tenga dichos péptidos en su composición. Se está probando una vacuna la ABvac40, como preventiva contra el alzhéimer desde el 2014. Su objetivo, es detener la producción de placas amiloides. La vacuna produciría anticuerpos encargados de eliminar los beta amiloides 40 y 42, que son los causantes de la neurodegeneración cerebral. Los ensayos de la vacuna se realizarán sobre un total de 24 personas: 16 pacientes diagnosticados y en estadio leve y ocho pacientes que reciben placebo. De acreditarse su inocuidad, la vacuna no estará en el mercado antes del 2018.
Ultrasonido
En marzo de 2015 se publicó en Science-Traslational Medicine una aproximación completamente nueva al tema alzhéimer, que ha sido probada en ratones. La misma utiliza una manera particular de aplicar ultrasonido, dentro del tejido gris cerebral. Estas ondas de sonido resultaron capaces de abrir gentilmente la barrera hematoencefálica, que separa al cerebro de la sangre, y estimularon a las células del Rio Ortega o microglia. Estas células microgliales, una vez activadas, resultaron capaces de ir desintegrando y eliminar las aglutinaciones beta-amiloideas del Alzheimer. Los autores de la investigación informaron haber observado la restauración completa de las memorias en el 75% de los ratones en los que ensayaron. Hallaron que los ratones así tratados desplegaron mejoras de la memoria en tres pruebas específicas. El equipo científico planea iniciar pruebas con animales de laboratorio superiores, como ovejas y simios, y espera ser autorizado a poner en marcha ensayos sobre seres humanos en el 2017.
Por su parte, en las aproximaciones clínicas clásicas (farmacológicas), un estudio de 2014 afirmaba que en ratones el antidepresivo citalopram detuvo el crecimiento de las placas beta amiloides de la enfermedad de Alzheimer existentes y redujo la formación de nuevas placas en un 78%. En un segundo experimento, los científicos administraron una dosis única de citalopram a 23 personas de entre 18 y 50 años que no estaban cognitivamente deterioradas ni padecían depresión. Cuando obtuvieron muestras de líquido cefalorraquídeo a las 24 horas, observaron una reducción del 37% en la producción de la proteína beta-amiloide.
Si esta enfermedad está relacionada con la resistencia a la insulina, se presentan múltiples alternativas terapéuticas. Se está evaluando actualmente el uso de medicamentos empleados en el tratamiento de la diabetes. Estudios recientes muestran que la administración de insulina por vía intranasal mejora la función cognitiva de pacientes normales y con alzhéimer. Una revisión sistemática de los ensayos clínicos hasta ahora desarrollados muestra resultados esperanzadores. Por otra parte, se ha propuesto el empleo de técnicas de inducción enzimática, con enzimas activas por la insulina.
Células madre
Otra de las áreas de investigación es la medicina regenerativa. Se trata de inyectar en el cerebro del paciente células madre embrionarias o adultas para intentar detener el deterioro cognitivo. Ya se han hecho experimentos en humanos con resultados positivos.
Marcapasos cerebral
La estimulación cerebral intenta normalizar la actividad, con un dispositivo llamado neuroestimulador, similar a un marcapasos cardíaco. El dispositivo forma parte de un tratamiento llamado estimulación cerebral profunda (ECP), que involucra la liberación de impulsos eléctricos para regular la actividad cerebral. La investigación, llevada a cabo en la Escuela de Medicina Johns Hopkins, forma parte de un proyecto más amplio iniciado en Canadá, donde ya se implantó el marcapasos a seis pacientes con la enfermedad. El tratamiento logró que los pacientes -todos con formas moderadas de alzhéimer- mostraran un incremento en la actividad neuronal durante 13 meses.
La terapia de estimulación cerebral profunda se ha utilizado con personas que sufren la enfermedad de Parkinson. Ahora, la terapia podría ser una alternativa para revertir el deterioro cognitivo de las personas con alzhéimer. Esta neurocirugía funcional, busca reparar, modular o corregir un déficit en un sistema o red neurológica determinada. Lo que ocurre con el alzhéimer es que se altera la química cerebral y esto conduce a una actividad eléctrica anormal que puede expresarse en temblores, deterioro cognitivo o trastornos psiquiátricos. La aplicación para el alzhéimer todavía está en sus primeras etapas.
Prevención
Ciertas actividades intelectuales, tales como el jugar al ajedrez, así como las interacciones sociales regulares, han sido asociadas en estudios epidemiológicos con un reducido riesgo de contraer la enfermedad de Alzheimer. Sin embargo, no se ha encontrado aún una relación causal.
Los estudios globales sobre las diferentes medidas que se pueden tomar para prevenir o retardar la aparición de la enfermedad de Alzheimer han tenido resultados contradictorios y no se ha comprobado aún una relación causal entre los factores de riesgo y la enfermedad, ni se han atribuido a efectos secundarios específicos. Por el momento, no parece haber medidas definitivas para prevenir la aparición del alzhéimer.
Varios estudios epidemiológicos han propuesto diversas relaciones entre ciertos factores modificables, tales como la dieta, los riesgos cardiovasculares, productos farmacéuticos o las actividades intelectuales entre otros, y la probabilidad de que en una población aparezca el alzhéimer. Por ahora se necesitan más investigaciones y ensayos clínicos para comprobar si estos factores ayudan a prevenirla.
La dieta mediterránea se ha asociado con un menor riesgo de desarrollar la enfermedad de Alzheimer, por su papel en la prevención del desarrollo de enfermedades cardiovasculares y por sus efectos antiinflamatorios y antioxidantes.
La curcumina del curry ha mostrado en estudios del 2001 y 2007 cierta eficacia en la prevención de daño cerebral en modelos de ratón.

Similares propiedades se comunicaron en 2010 y 2012 para ensayos en ratones de la Withania somnifera (ashwagandha, ginseng indio).
A pesar de que los riesgos cardiovasculares, como la hipercoleterolemia, hipertensión arterial, la diabetes y el tabaquismo, están asociados a un mayor riesgo de desarrollo y progresión del alzhéimer, las estatinas, que son medicamentos que disminuyen la concentración de colesterol en el plasma sanguineo, no han sido efectivas en la prevención o mejoramiento del alzhéimer. Sin embargo, en algunos individuos, el uso a largo plazo de los antiinflamatorios no esteroideos (AINEs) está vinculado con una reducción de la probabilidad de padecerla. Otros fármacos y terapias, como el reemplazo de hormonas en las mujeres, han dejado de ser aconsejadas como medidas preventivas del alzhéimer. Se incluye también un reporte en 2007 que concluyó la falta de evidencias significativas y la presencia de inconsistencias en el uso de ginkgo biloba para mejorar los trastornos cognitivos.
Hay diferentes actividades intelectuales, como el jugar al ajedrez, la lectura, el completar crucigramas o las interacciones sociales frecuentes, que parecen retardar la aparición y reducir la gravedad del alzhéimer. El hablar varios idiomas también parece estar vinculado a la aparición tardía de la enfermedad.
Otros estudios han sugerido un posible aumento en el riesgo de la aparición del alzhéimer con la exposición a campos magnéticos, la ingestión de metales, en particular de aluminio, o la exposición a ciertos solventes. La calidad de algunos de estos estudios ha sido criticada, y otros estudios han concluido que no hay una relación entre estos factores ambientales y la aparición del alzhéimer.
Algunas asociaciones científicas actualmente trabajan en el campo médico y social recomendando y/o asesorando a las familias de los pacientes, propiciando a su vez la investigación, tales como Alzheimer’s Asociation y Alzheimer Argentina.
ARTICULO DEL PERIODICO: LA AUTOESTIMA

LA AUTOESTIMA

Todos tenemos una imagen mental de quienes somos, qué aspecto tenemos, en qué somos buenos y cuáles son nuestros puntos débiles. Nos formamos esa imagen a lo largo del tiempo, empezando en nuestra infancia. El término auto-imagen se utiliza para referirse a la imagen mental que una persona tiene de sí. Gran parte de nuestra auto-imagen se basa en nuestras interacciones con otras personas y nuestras experiencias vitales. Esta imagen mental (nuestra auto-imagen) contribuye a nuestra autoestima.

En virtud de este razonamiento, incluso los seres humanos más viles merecen un trato humano y considerado. Esta actitud, no obstante, no busca entrar en conflicto con los mecanismos que la sociedad tenga a su disposición para evitar que unos individuos causen daño a otros—sea del tipo que sea.

El concepto de autoestima varía en función del paradigma psicológico que lo aborde (psicología humanista, psicoanalisis o conductismo). Desde el punto de vista del psicoanálisis, la autoestima está íntimamente relacionada con el desarrollo del ego ; por otro lado, el conductismo se centra en conceptos tales como estímulo, respuesta, refuerzo y aprendizaje, con lo cual el concepto holístico de autoestima no tiene sentido. La autoestima es además un concepto que ha traspasado frecuentemente el ámbito exclusivamente científico para formar parte del lenguaje popular. El budismo considera al ego una ilusión de la mente, de tal modo que la autoestima, e incluso el alma, son también ilusiones; el amor y la compasión hacia todos los seres con sentimientos y la nula consideración del ego, constituyen la base de la felicidad absoluta. En palabras de Buda, «no hay un camino hacia la felicidad, la felicidad es el camino”

Según la Organización Mundial de la Salud, La autoestima es un conjunto de percepciones, pensamientos, evaluaciones, sentimientos y tendencias de comportamiento dirigidas hacia uno mismo, hacia nuestra manera de ser, y hacia los rasgos de nuestro cuerpo y nuestro carácter. En resumen: es la evaluación perceptiva de nosotros mismos. En su jerarquía de las necesidades humanas, se describe como la necesidad de aprecio, que se divide en dos aspectos, el que se tiene uno mismo (amor propio, confianza, pericia, suficiencia, etc.), y el respeto y estimación que se recibe de otras personas (reconocimiento, aceptación, etc.). La expresión de aprecio más sana según Maslow es la que se manifiesta «en el respeto que le merecemos a otros, más que el renombre, la celebridad y la adulación».

La capacidad de desarrollar una autoestima y un respeto saludable por sí mismo es propia de la naturaleza de los seres humanos, ya que el solo hecho de poder pensamiento constituye la base de suficiencia, y el único hecho de estar vivos es la base de su derecho a esforzarse por conseguir felicidad. Así pues, el estado natural del ser humano debería corresponder a una autoestima alta. Sin embargo, la realidad es que existen muchas personas que, lo reconozcan o no, lo admitan o no, tienen un nivel de autoestima inferior al teóricamente natural.

Ello se debe a que, a lo largo del desarrollo, y a lo largo de la vida en sí, las personas tienden a apartarse de la auto-conceptualización [y conceptualización] positiva, o bien a no acercarse nunca a ellas; los motivos por los que esto ocurre son diversos, y pueden encontrarse en la influencia negativa de otras personas, en un auto-castigo por haber faltado a los valores propios [o a los valores de su grupo social], o en un déficit de comprensión de compasión por las acciones que uno realiza. [y, por extensión, de las acciones que realizan los demás].

La autoestima es un concepto gradual. En virtud de ello, las personas pueden presentar en esencia uno de tres estados:
• Tener una autoestima alta equivale a sentirse confiadamente apto para la vida, o, usando los términos de la definición inicial, sentirse capaz y valioso; o sentirse aceptado como persona.

• Tener una autoestima baja es cuando la persona no se siente en disposición para la vida; sentirse equivocado como persona.

• Tener un término medio de autoestima es oscilar entre los dos estados anteriores, es decir, sentirse apto e inútil, acertado y equivocado como persona, y manifestar estas incongruencias en la conducta—actuar, unas veces, con sensatez, otras, con irreflexión—-, reforzando, así, la inseguridad.

En la práctica, y según la experiencia de Nathaniel Branden, todas las personas son capaces de desarrollar la autoestima positiva, al tiempo que nadie presenta una autoestima totalmente sin desarrollar. Cuanto más flexible es la persona, tanto mejor resiste todo aquello que, de otra forma, la haría caer en la derrota o la desesperación.
Existe una escalera de la autoestima que va desde el punto mas bajo (auto reconocimiento hasta el punto mas alto que es la autosuperación.

Autoreconocimiento: Es reconocerse a sí mismo, reconocer las necesidades, habilidades, potencialidades y debilidades, cualidades corporales o psicológicas, observar sus acciones, como actúa, por qué actúa y qué siente.

Autoaceptación: Es la capacidad que tiene el ser humano de aceptarse tal como es, en lo físico, psicológico y social; aceptar cómo es su conducta consigo mismo y con los otros. Es admitir y reconocer todas las partes de sí mismo como un hecho, como forma de ser y sentir.

Autovaloración: Refleja la capacidad de evaluar y valorar las cosas que son buenas de uno mismo, aquellas que le satisfacen y son enriquecedoras, le hacen sentir bien, le permiten crecer y aprender. Es buscar y valorar todo aquello que le haga sentirse orgulloso de sí mismo.

Autorrespeto: Expresar y manejar en forma conveniente sentimientos y emociones, sin hacerse daño ni culparse. El respeto por sí mismo es la sensación de considerarse merecedor de la felicidad, es tratarse de la mejor forma posible, no permitir que los demás lo traten mal; es el convencimiento real de que los deseos y las necesidades de cada uno son derechos naturales, lo que permitirá poder respetar a los otros con sus propias individualidades.

Autosuperación: Si la persona se conoce es consciente de sus cambios, crea su propia escala de valores, desarrolla y fortalece sus capacidades y potencialidades, se acepta y se respeta; está siempre en constante superación, por lo tanto, tendrá un buen nivel de autoestima, generando la capacidad para pensar y entender, para generar, elegir y tomar decisiones y resolver asuntos de la vida cotidiana, escuela, amigos, familia, etc. Es una suma de pequeños logros diarios.

La autoestima es uno de los temas mas trabajados en las sesiones de tratamiento psicológico y/o psiquiatrico.

La autoestima permite a las personas enfrentarse a la vida con mayor confianza, benevolencia y optimismo, y por consiguiente alcanzar más fácilmente sus objetivos y autorrealizarse .

Permite que uno sea más ambicioso respecto a lo que espera experimentar emocional, creativa y espiritualmente. Desarrollar la autoestima es ampliar la capacidad de ser felices; la autoestima permite tener el convencimiento de merecer la felicidad.

Comprender esto es fundamental, y redunda en beneficio de todos, pues el desarrollo de la autoestima positiva aumenta la capacidad de tratar a los demás con respeto, benevolencia y buena voluntad, favoreciendo así las relaciones interpersonales enriquecedoras y evitando las destructivas.

Permite la creatividad en el trabajo, y constituye una condición especialmente crítica para la profesión docente.

Rosenberg entiende a la autoestima como un fenómeno actitudinal creado por fuerzas sociales y culturales. La autoestima se crea en un proceso de comparación que involucra valores y discrepancias. El nivel de autoestima de las personas se relaciona con la percepción del sí mismo en comparación con los valores personales. Estos valores fundamentales han sido desarrollados a través del proceso de socialización. En la medida que la distancia entre el sí mismo ideal y el sí mismo real es pequeña, la autoestima es mayor. Por el contrario, cuanto mayor es la distancia, menor será la autoestima, aun cuando la persona sea vista positivamente por otros.
La autoestima es un constructo de gran interés clínico por su relevancia en los diversos cuadros psicopatológicos, así como por su asociación con la conducta de búsqueda de ayuda psicológica, con el estrés y con el bienestar general. (Vázquez,Jiménez & Vázquez, 2004.)

Muy particularmente se ha asociado con cuadros como la depresión, los trastornos alimentarios, los trastornos de personalidad, la ansiedad, y la fobia social. Asimismo se ha señalado que el nivel de autoestima es un excelente predictor de la depresión.
El estudio de la autoestima es, por tanto, un aspecto esencial en la investigación psicopatológica, siendo de interés la disponibilidad de instrumentos adecuadamente validados para su evaluación.

La Escala de Autoestima de Rosenberg es una de las escalas más utilizadas para la medición global de la autoestima. Desarrollada originalmente por Rosenberg (1965) para la evaluación de la autoestima en adolescentes, incluye diez ítems cuyos contenidos se centran en los sentimientos de respeto y aceptación de sí mismo/a. La mitad de los ítems están enunciados positivamente y la otra mitad negativamente (ejemplos, sentimiento positivo: » creo que tengo un buen número de cualidades » sentimiento negativo: » siento que no tengo muchos motivos para sentirme orgulloso de mi» ). Es un instrumento unidimensional que se contesta en una escala de 4 alternativas, que va desde » muy de acuerdo» a » muy en desacuerdo”.

Siempre nos encontramos con falsos estereotipos como:
1.- La comodidad no es autoestima. A una persona con la autoestima baja —o «equivocada», según la terminología de Branden—, cualquier estímulo positivo, a lo más que podrá llegar, será a hacerla sentir cómoda o, a lo sumo, mejor con respecto a sí misma únicamente durante un tiempo. Por lo tanto, los bienes materiales, o las relaciones sexuales, o el éxito, o el aspecto físico, por sí solos, producirán sobre esa persona comodidad, o bien un falso y efímero desarrollo de la autoestima, pero no potenciarán realmente la confianza y el respeto hacia uno mismo.

2.- La autoestima no es competitiva ni comparativa. Paradójicamente, la mayoría de las personas buscan la autoconfianza y el autorrespeto fuera de sí mismas, motivo por el cual están abocadas al fracaso. Según Nathaniel Branden, «la autoestima se comprende mejor como una suerte de logro espiritual o mental, es decir, como una victoria en la evolución de la conciencia». Así, la autoestima proporciona serenidad espiritual, la cual a su vez permite a las personas disfrutar de la vida.
La verdadera autoestima no se expresa mediante la autoglorificación a expensas de los demás, o por medio del afán de ser superior a otras personas o de rebajarlas para elevarse uno mismo. La arrogancia, la jactancia y la sobrevaloración de las propias capacidades revelan una autoestima equivocada, y no un exceso de autoestima. La autoestima es la base fundamental para que el ser humano desarrolle al máximo sus capacidades, es el punto de partida para el desarrollo positivo de las relaciones humanas, del aprendizaje, de la creatividad y de la responsabilidad personal.

La persona que se autoestima suficientemente:
1. Cree con firmeza en ciertos valores y principios, y está dispuesta a defenderlos incluso aunque encuentre oposición. Además, se siente lo suficientemente segura de sí misma como para modificarlos si la experiencia le demuestra que estaba equivocada.

2. Es capaz de obrar según crea más acertado, confiando en su propio criterio, y sin sentirse culpable cuando a otros no les parezca bien su proceder.

3. No pierde el tiempo preocupándose en exceso por lo que le haya ocurrido en el pasado ni por lo que le pueda ocurrir en el futuro. Aprende del pasado y proyecta para el futuro, pero vive con intensidad el presente.

4. Confía plenamente en su capacidad para resolver sus propios problemas, sin dejarse acobardar fácilmente por fracasos y dificultades. Y, cuando realmente lo necesita, está dispuesta a pedir la ayuda de otros.

5. Como persona, se considera y siente igual que cualquier otro; ni inferior, ni superior; sencillamente, igual en dignidad; y reconoce diferencias en talentos específicos, prestigio profesional o posición económica.

6. Da por sentado que es interesante y valiosa para otras personas, al menos para aquellos con los que mantiene amistad.

7. No se deja manipular, aunque está dispuesta a colaborar si le parece apropiado y conveniente.

8. Reconoce y acepta en sí misma diferentes sentimientos y pulsiones, tanto positivos como negativos, y está dispuesta a revelárselos a otra persona, si le parece que vale la pena y así lo desea.

9. Es capaz de disfrutar con una gran variedad de actividades.

10. Es sensible a los sentimientos y necesidades de los demás; respeta las normas sensatas de convivencia generalmente aceptadas, y entiende que no tiene derecho —ni lo desea— a medrar o divertirse a costa de otros.

Cuando alguien visita a un psicólogo es porque no cuenta con las herramientas suficientes para solucionar un problema. Nosotros les proporcionamos esas herramientas para que el paciente pueda utilizarlas como mejor pueda.

La idea que la mayoría de la población tiene de lo que hacemos los psicólogos es equivocada. Un psicólogo no da consejos ni ordenes solo le da varias alternativas o herramientas para que las utilice el paciente en beneficio propio.

Una de estas herramientas y la fundamental es la autoestima. Cuando tenemos una alta autoestima somos capaces de enfrentarnos a cualquier problema, situación o circunstancia que la vida nos proponga. Es por ello que en PSICOLOGIA SIN LIMITACIONES lo primero que hacemos es trabajar la autoestima.

PAREJAS EN LA WEB

DISCAPACIDAD SENSORIAL

Los tipos de discapacidad sensorial no incapacitan a la persona para tener una vida funcional en la mayoría de los aspectos si lleva a cabo un adecuado adiestramiento. Casos bien conocidos, como Hellen Keller, (oradora y activista política, primera persona sordociega en conseguir una licenciatura universitaria) demostraron que la incapacidad sensorial no es obstáculo para el desarrollo de una vida funcional y exitosa.
Discapacidad sensorial
La discapacidad sensorial corresponde a las personas con deficiencias visuales y auditivas, quienes presentan problemas en la comunicación y el lenguaje. Los juegos y juguetes educativos forman parte primordial del proceso de aprendizaje infantil, sobre todo en niños afectados por algún tipo de discapacidad. Los juegos son un recurso imprescindible para quienes se ven afectados por lesiones o capacidades diferentes, pues siguen siendo niños, y el juego es una necesidad para el desarrollo infantil en todos los aspectos. Akros Educational ofrece los juegos infantiles adaptados a las más diversas necesidades de cada niño, según la categoría de la discapacidad.

Discapacidad sensorial es la pérdida o atenuación de una o más funciones sensoriales humanas: la auditiva, visual o ambas. Su presencia no afecta de ninguna manera en el potencial muscular y funcional del pequeño, sino su vida social. Las discapacidades sensoriales a menudo afectan más allá de la capacidad de comunicación, también a la autoimagen de la persona y a su desempeño en la vida cotidiana. Esta nomenclatura se refiere a tres tipos de discapacidad:
• Ceguera o hipovisión con agudeza visual no superior a 3/10.
• Sordera o pérdida auditiva mayor de 25 decibelios en ambos oídos.
• Sordoceguera se caracteriza por la coexistencia de ambos discapacidades sensoriales visuales y auditivas.

Definición de discapacidad visual
Podemos considerar la existencia de discapacidad visual en aquellas personas completamente ciegas o con un resto de visión tan pequeño que no se puede corregir con lentes normales. Las causas de discapacidad visual pueden ser, entre otras, factores genéticos, las alteraciones visuales, consecuencia de cierta medicación, o contraer la rubéola durante el embarazo. Es importante distinguir entre una situación de deficiencia visual severa que se establece en el nacimiento y la ceguera que afecta en la edad adulta. Las repercusiones psicológicas y adaptaciones a llevar a cabo son completamente diferentes.

La deficiencia visual en la infancia
Las consecuencia más evidente que afecta al desarrollo de un niño ciego es la ralentización en el desarrollo psicomotor. El niño más adelante aprende a usar sus otros sentidos (oído y el tacto) para reconstruir mentalmente la forma y la ubicación de los objetos en el espacio. En una persona ciega, el proceso de conocimiento del mundo y de las relaciones sociales resulta diferente al de una persona que ve normalmente, pues tiene que confiar completamente en el lenguaje verbal.

La sobreprotección de la familia a menudo puede prevenir la independencia y la falta de expansión de las relaciones sociales. Para evitarla también se necesita brindar apoyo y asesoramiento psicológico a la familia del niño, para que aprendan a exponer al niño a diferentes contextos sociales, e irlos ampliando progresivamente. Si el apoyo es el adecuado desde la infancia, el niño ciego es capaz de integrarse plenamente en la sociedad, asumiendo un papel destacado en lo personal y en lo laboral.

La deficiencia visual en la edad adulta
El principal problema que surge cuando una persona pierde la vista en una edad más adulta consiste en la negación de la realidad de la enfermedad. El defecto visual presente en el nacimiento estimula otros órganos de los sentidos, pero si el daño se produce en la edad adulta la persona ya ha establecido un modo de conocimiento basado en el uso de la visión y el déficit repentino o progresiva puede ser compensado en menor medida. La persona percibe la pérdida de la visión con una intensidad dolorosa y todos los fenómenos relacionados con el duelo se producen fuertemente.

Discapacidad en la audición
En la discapacidad auditiva se dañan los sistemas que permiten recibir las ondas de sonido. Si no se reciben, no se pueden transformar en impulsos nerviosos y se imposibilita la posterior transmisión de tales impulsos a la parte de la corteza cerebral que los procesa. La pérdida de la audición o sordera puede ser de dos tipos en relación con las estructuras dañadas.

La pérdida de audición conductiva es causada generalmente por anomalías en el oído externo o el oído medio e implica una pérdida de la audición uniforme. La pérdida de la audición perceptiva es en cambio causada por el daño al sistema que transforma el pulso de sonido en un impulso nervioso, en estos casos, la pérdida de audición es mayor para las frecuencias altas. Existen baremos para marcar distintos grados de discapacidad sensorial, por ejemplo en el caso de la discapacidad auditiva (hipoacusia) existen los siguientes:
• Audición normal. La persona es capaz de percibir sonidos entre 10 y 15 dB. (Ruido al respirar).
• Audición limitada. El rango de percepción es menor y queda en valores entre los 16 y los 25 dB. (Sonido en una biblioteca con gente).
• Pérdida superficial. La persona oye entre 26 y 40 dB. (Volumen normal de una conversación).
• Pérdida moderada entre 41 y 55 dB. (Aglomeración de personas).
• Pérdida moderada a severa. Se escuchan sonidos entre 56 y 70 dB. (Aspiradora)
• Pérdida severa. La persona percibe sonidos a partir de 71 hasta 90 dB. (Tren)
• Pérdida profunda. La persona es incapaz de percibir sonidos de más de 90 dB.

Discapacidad auditiva en niños
La consecuencia más importante de pérdida de la audición o sordera, debido a la percepción de difícil o imposible de sonidos, consiste en la percepción limitada de la lengua y cómo afecta a la rapidez del desarrollo intelectual. El lenguaje, además de tener una función comunicativa, desempeña tiene una función reguladora del comportamiento. Esta situación puede resolverse mediante el lenguaje de signos, que ofrece la oportunidad de mejorar el rendimiento académico.

La pérdida de audición en un bebé también se deja notar en términos del desarrollo emocional y social. Los recién nacidos reaccionan selectivamente a la voz de la madre y, como los niños sordos no son capaces de hacer eso, afecta al establecimiento del proceso de unión.
La pérdida de audición leve puede ser compensada de manera efectiva sin afectar el desarrollo. Los niños que nacen con pérdida de audición leve pueden desarrollar el lenguaje como los demás niños. Tienen dificultades para entender el significado de las palabras, a y a veces puede confundir a algunas letras y fonemas.

Las familias y amigos de niños con discapacidades auditivas de nivel medio pueden aumentar el volumen de la voz en presencia del niño, pero deben asumir que precisa una terapia del lenguaje obligatoriamente, y puede que una prótesis.
Las personas con pérdida de audición severa no perciben ningún sonido del habla. Si se les habla con voz muy alta pueden recibir algún sonido y su ritmo. Sólo una intervención de terapia del habla puede ayudarlo a aprender a comunicarse. Muchas personas con sordera total pueden, además de leer los labios, hablar mediante el control de su voz a través de la vibración de las cuerdas vocales, e incluso cantar.
Los casos más graves son aquellos en los que la discapacidad auditiva se asocia con lo visual pero, como se nombró al principio, no resulta tan limitante como pudiera parecer si la persona recibe un adecuado entrenamiento.
